Processing Samples with pfhrp3 13-5++ deletion pattern
Setup
Downloads
Code
allMetaSamplesByBio = readr::read_tsv("../../meta/metadata/metaByBioSample.tab.txt")
allMetaSamples = readr::read_tsv("../../meta/metadata/meta.tab.txt")
allMetaDeletionCalls = readr::read_tsv("../metaSelected.tab.txt")
allMetaDeletionCalls_possiblyHRP2Deleted = allMetaDeletionCalls %>%
filter(possiblyHRP2Deleted)
allMetaDeletionCalls_possiblyChr11Deleted = allMetaDeletionCalls %>%
filter(possiblyChr11Deleted)
allMetaDeletionCalls_Hrp3pattern2 = readr::read_tsv("../allMetaDeletionCalls_Hrp3pattern2.tab.txt")
hrp3Pat2_samplesWithTare1 = read_tsv("hrp3Pat2_samplesWithTare1.tsv")
realmccoilCoiCalls = readr::read_tsv("../wgs_variants/THEREALMcCOIL/categorical_method/real_mccoil_COI_calls.tsv")
realmccoilCoiCalls_poly = realmccoilCoiCalls %>%
filter(random_median != 1 | topHE_median != 1) %>%
left_join(allMetaSamples %>%
select(sample, BiologicalSample))
# PH0681-C and PV0257-C were mixed Investigating pfmdr1 duplication
Regions around where there appears to be transition
testRegion.bed
Pf3D7_13_v3 2835411 2835511 Pf3D7_13_v3-2835411-2835511-for 100 +
Pf3D7_13_v3 2835461 2835561 Pf3D7_13_v3-2835461-2835561-for 100 +
Pf3D7_13_v3 2835511 2835611 Pf3D7_13_v3-2835511-2835611-for 100 +
Pf3D7_13_v3 2835561 2835661 Pf3D7_13_v3-2835561-2835661-for 100 +
Pf3D7_13_v3 2835611 2835711 Pf3D7_13_v3-2835611-2835711-for 100 +
Pf3D7_13_v3 2835661 2835761 Pf3D7_13_v3-2835661-2835761-for 100 +
Get cross mapping sequences
Code
cd /tank/data/plasmodium/falciparum/pfdata/hrp3_deletion_MDR1_dup_investigation
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-093.sorted.bam --bed testRegion.bed | egrep Pf3D7_05_v3 | cut -f16-21 | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN > IGS-CBD-093_transpositionPoint.bed
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-060.sorted.bam --bed testRegion.bed | egrep Pf3D7_05_v3 | cut -f16-21 | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN > IGS-CBD-060_transpositionPoint.bed
for x in `cat /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt`; do elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/${x}.sorted.bam --bed testRegion.bed | egrep Pf3D7_05_v3 | cut -f16-21 | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN > ${x}_transpositionPoint.bed ; done;
elucidator createBedRegionFromName --names Pf3D7_05_v3-978580-979265 > Pf3D7_05_v3-978580-979265.bed
#cat *_transpositionPoint.bed | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN | egrep Pf3D7_05_v3-978580-979265 > Pf3D7_05_v3-978580-979265.bed
elucidator extendBedRegions --bed Pf3D7_05_v3-978580-979265.bed --left 1000 --right 10000 > Pf3D7_05_v3-978580-979265.bed_l1000_r10000.bed
elucidator createWindowsInRegions --bed Pf3D7_05_v3-978580-979265.bed_l1000_r10000.bed --step 50 --windowSize 100 --minLen 50 > Pf3D7_05_v3-978580-979265.bed_l1000_r10000_wstep50_wsize100.bed
rm -fr intersectWith_Pf3D7_05_v3-978580-979265.txt && for x in *_transpositionPoint.bed; do echo -e ${x} $(elucidator getOverlappingBedRegions --bed ${x} --intersectWithBed Pf3D7_05_v3-978580-979265.bed) >> intersectWith_Pf3D7_05_v3-978580-979265.txt ; done;samplesWithTransitionToChr5
Code
samplesWithTransitionToChr5 = tibble(
sample =
c(
"IGS-CBD-015",
"IGS-CBD-020",
"IGS-CBD-036",
"IGS-CBD-044",
"IGS-CBD-060",
"IGS-CBD-093",
"IGS-CBD-122",
"IVC11-BB-0084",
"PH0143-CW",
"PH0229-C",
"PH0274-C",
"PH0278-CW",
"PH0308-CW",
"PH0397-C",
"PH0413-C",
"PH0416-C",
"PH0457-CW",
"PH0467-CW",
"PH0474-CW",
"PH0480-C",
"PH0529-C",
"PH0534-C",
"PH0544-C",
"PH0697-C",
"PH0817-C",
"PH0894-C",
"PH0959-Cx",
"PV0194-C",
"SPT24457"
)
)
samplesWithTransitionToChr5 = samplesWithTransitionToChr5 %>%
left_join(allMetaDeletionCalls)
create_dt(samplesWithTransitionToChr5)Code
write_tsv(samplesWithTransitionToChr5, "samplesWithTransitionToChr5.tsv")Code
nohup elucidator runMultipleCommands --cmdFile runRegionBamTrimCmds.txt --additionalFields "{DIRNAME}:Pf3D7_05_v3-978580-979265_l1000_r10000_wstep50_wsize100;{SAMPLE}:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/pfdata/finalHRPII_HRPIII_windows_investTelemeraseHealing/Pf3D7_05_v3-978580-979265.bed_l1000_r10000_wstep50_wsize100.bed\";{NCPUS}:4 --keepTemporaryFiles --qualCheck 5 --keepImproperMates" --replaceFields --numThreads 11 &
cd Pf3D7_05_v3-978580-979265_l1000_r10000_wstep50_wsize100
PathWeaver runProcessClustersOnRecon --overWriteDir --dout popClustering --pat _Pf3D7_05_v3-978580-979265_l1000_r10000_wstep50_wsize100 --numThreads 5 --groupingsFile /tank/data/plasmodium/falciparum/pfdata/metadata/meta.tab.txt --samples /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt Code
cd /tank/data/plasmodium/falciparum/pfdata/hrp3_deletion_MDR1_dup_investigation
for x in `cat /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt`; do elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/${x}.sorted.bam --bed Pf3D7_05_v3-978580-979265.bed | egrep Pf3D7_13_v3 | cut -f16-21 | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN > ${x}_transpositionPoint_reverseTo13.bed ; done;
cat *_transpositionPoint_reverseTo13.bed | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN | egrep Pf3D7_13_v3-2835112-2835673 > Pf3D7_13_v3-2835112-2835673.bed
cat Pf3D7_13_v3-2835112-2835673.bed Pf3D7_05_v3-978580-979265.bed | sed 's/Pf3D7_05_v3-978580-979265/Pf3D7_13_v3-2835112-2835673/g' > combined_Pf3D7_05_v3-978580-979265_Pf3D7_13_v3-2835112-2835673.bed
# make the regions opposite directions
nohup elucidator runMultipleCommands --cmdFile runRegionCmds.txt --additionalFields "{DIRNAME}:combined_Pf3D7_05_v3-978580-979265_Pf3D7_13_v3-2835112-2835673;{SAMPLE}:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/pfdata/finalHRPII_HRPIII_windows_investTelemeraseHealing/combined_Pf3D7_05_v3-978580-979265_Pf3D7_13_v3-2835112-2835673.bed\";{NCPUS}:4 --keepTemporaryFiles --qualCheck 5 --keepImproperMates" --replaceFields --numThreads 11 &Code
elucidator extractFromGenomesAndCompare --genomeDir /tank/data/genomes/plasmodium//genomes/pf/genomes/ --gffDir /tank/data/genomes/plasmodium//genomes/pf/info/gff/ --numThreads 14 --target combined_Pf3D7_05_v3-978580-979265_Pf3D7_13_v3-2835112-2835673 --program PathWeaver --sample bottomLevel --verbose --overWriteDir --dout compToAll_bottomLevel --fasta bottomLevel.fasta
elucidator extractFromGenomesAndCompare --genomeDir /tank/data/genomes/plasmodium//genomes/pf/genomes/ --gffDir /tank/data/genomes/plasmodium//genomes/pf/info/gff/ --numThreads 14 --target combined_Pf3D7_05_v3-978580-979265_Pf3D7_13_v3-2835112-2835673 --program PathWeaver --sample largeInsert --verbose --overWriteDir --dout compToAll_largeInsert --fasta largeInsert.fasta
elucidator extractFromGenomesAndCompare --genomeDir /tank/data/genomes/plasmodium//genomes/pf/genomes/ --gffDir /tank/data/genomes/plasmodium//genomes/pf/info/gff/ --numThreads 14 --target combined_Pf3D7_05_v3-978580-979265_Pf3D7_13_v3-2835112-2835673 --program PathWeaver --sample topLevel --verbose --overWriteDir --dout compToAll_topLevel --fasta topLevel.fasta
elucidator trimToLen --fasta largeInsert.fasta --length 530
elucidator extractFromGenomesAndCompare --genomeDir /tank/data/genomes/plasmodium//genomes/pf/genomes/ --gffDir /tank/data/genomes/plasmodium//genomes/pf/info/gff/ --numThreads 14 --target combined_Pf3D7_05_v3-978580-979265_Pf3D7_13_v3-2835112-2835673 --program PathWeaver --sample largeInsert --verbose --overWriteDir --dout compToAll_trimmed_largeInsert --fasta trimmed_largeInsert.fasta
elucidator extractFromGenomesAndCompare --genomeDir /tank/data/genomes/plasmodium//genomes/pf/genomes/ --gffDir /tank/data/genomes/plasmodium//genomes/pf/info/gff/ --numThreads 14 --target combined_Pf3D7_05_v3-978580-979265_Pf3D7_13_v3-2835112-2835673 --program PathWeaver --sample largeInsert --verbose --overWriteDir --dout compToPf3D7_trimmed_largeInsert --genomes Pf3D7 --fasta trimmed_largeInsert.fasta
elucidator trimFront --fasta largeInsert.fasta --forwardBases 530 --overWrite --out trimmedFront_largeInsert
elucidator extractFromGenomesAndCompare --genomeDir /tank/data/genomes/plasmodium//genomes/pf/genomes/ --gffDir /tank/data/genomes/plasmodium//genomes/pf/info/gff/ --numThreads 14 --target combined_Pf3D7_05_v3-978580-979265_Pf3D7_13_v3-2835112-2835673 --program PathWeaver --sample largeInsert --verbose --overWriteDir --dout compToAll_trimmedFront_largeInsert --fasta trimmedFront_largeInsert.fasta
elucidator extractFromGenomesAndCompare --genomeDir /tank/data/genomes/plasmodium//genomes/pf/genomes/ --gffDir /tank/data/genomes/plasmodium//genomes/pf/info/gff/ --numThreads 14 --target combined_Pf3D7_05_v3-978580-979265_Pf3D7_13_v3-2835112-2835673 --program PathWeaver --sample largeInsert --verbose --overWriteDir --dout compToPf3D7_trimmedFront_largeInsert --genomes Pf3D7 --fasta trimmedFront_largeInsert.fastaPoint of on Chr05, start of [AT]+ 979203 (but then goes in the reverse complement direction), ends at 952574 (26629)
Point of on Chr13, start of [AT]+ 2835587
There is evidence of a genomic re-arrangement that involves an AT di-nucleotide repeat at 2835587 on chromosome 13 and an AT di-nucleotide repeat at 979203 on chromosome 5 as evidenced by paired-end reads with discordant mates with one mate mapping to chromosome 13 and one mapping to chromosome 5. When reads are extracted from these regions and assembled a contig goes up through 2835587 on chromosome 13 and then into the reverse strand on chromosome 5 at 979203. This likely causes the duplication of a 26K BP region of chromosome 5 starting at 979203 and going along the reverse strand to 952574 given the double of coverage of this region and that at the region where coverage normalizes again the tandem repeat associate element 1(TARE1)(Vernick and McCutchan 1988) is detected contiguous with the genomic sequence at position 952574, within the gene PF3D7_0522900 (a zinc finger gene). This results in the duplication of the following genes: PF3D7_0523600, PF3D7_0523500, PF3D7_0523400, PF3D7_0523300, PF3D7_0523200, PF3D7_0523100, PF3D7_0523000 (MDR1). This re-arrangement results in the deletion of pfhrp3 and the duplication of MRD1. The duplication of MDR1 could be the driver of this genomic re-arrangement event.
Code
# Pf3D7_05_v3-978580-979265MDR1 duplication
Narrow stable windows
These windows are non-paralogous regions without long tandem repeats within Pf3D7 which can be used to genotype this region.
Code
stableWindows = readr::read_tsv("~/Dropbox_Personal/ownCloud/documents/plasmodium/falciparum/newRedesignHeome_2021_04/Pf_Determine_Regions_of_Interest/windows/mergedFinalLargeWindows.bed", col_names = F)
stableWindows_05_regionOfInterestNarrow = stableWindows %>%
filter(X1 == "Pf3D7_05_v3",
X2 >= 978580 - 10000 * 5,
X2 <= 979265 + 10000)
write_tsv(stableWindows_05_regionOfInterestNarrow, "stableWindows_05_regionOfInterestNarrow.bed", col_names = F)Narrow finner windows
These windows are used to get a more refined map of coverage within the pfmdr1 region.
Code
stableWindows = readr::read_tsv("~/Dropbox_Personal/ownCloud/documents/plasmodium/falciparum/newRedesignHeome_2021_04/Pf_Determine_Regions_of_Interest/windows/mergedFinalLargeWindows.bed", col_names = F)
stableWindows_05_regionOfInterestNarrow_full = stableWindows %>%
filter(X1 == "Pf3D7_05_v3",
X2 >= 978580 - 10000 * 5,
X2 <= 979265 + 10000) %>%
group_by(X1) %>%
summarise(X2 = min(X2),
X3 = max(X3)) %>%
mutate(X4 = paste0(X1, "-", X2, "-", X3),
X5 = X3 - X2,
X6 = "+")
write_tsv(stableWindows_05_regionOfInterestNarrow_full, "stableWindows_05_regionOfInterestNarrow_full.bed", col_names = F)Code
elucidator createWindowsInRegions --bed stableWindows_05_regionOfInterestNarrow_full.bed --step 150 --windowSize 200 --minLen 50 | elucidator bedGetIntersectingGenesInGff --gff /tank/data/genomes/plasmodium/genomes/pf/info/gff/Pf3D7.gff --extraAttributes description --bed STDIN --overWrite --out stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200.bed
rsync -raPh stableWindows_05_regionOfInterestNarrow_full.bed hathawan@calderon.barrel-of-knowledge.info:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22
rsync -raPh stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200.bed hathawan@calderon.barrel-of-knowledge.info:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22Code
nohup elucidator runMultipleCommands --cmdFile runRegionBamTrimCmds.txt --additionalFields "{DIRNAME}:stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200;{SAMPLE}:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200.bed\";{NCPUS}:4 --keepTemporaryFiles --qualCheck 5 --keepImproperMates" --replaceFields --numThreads 11 &
nohup elucidator runMultipleCommands --cmdFile runRegionBamTrimCmds.txt --additionalFields "{DIRNAME}:stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200;{SAMPLE}:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_samples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200.bed\";{NCPUS}:4" --replaceFields --numThreads 11 &
cd stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200
PathWeaver runProcessClustersOnRecon --overWriteDir --dout popClustering --pat _stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200 --numThreads 5 --groupingsFile /tank/data/plasmodium/falciparum/pfdata/metadata/meta.tab.txt --samples /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_samples.txt
elucidator countPWExtractedReadsWithPattern --seqPat "TT[CT]AGGGTT[CT]AGGG","CCCT[AG]AACCCT[AG]AA" --pat _stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200 --bedFnp /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200.bed --minReadCounts 1 --numThreads 10 --overWrite --out stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_countTares.txt --samples /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txtCode
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_countTares = readr::read_tsv("stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_countTares.txt")
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_countTares_sum = stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_countTares %>%
group_by(sample, region) %>%
summarise(count = sum(count)) %>%
separate(region, convert = T, remove = F, sep = "-", into = c("chrom", "start", "end", "strand"))
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_countTares_sum_filt = stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_countTares_sum %>%
filter("Pf3D7_05_v3-952484-952684-for" == region)TARE1 sequence begins at in the reverse strand direction Pf3D7_05_v3 952668 (not inclusive, 952667 is the first base where the TARE1 )
investigating additional copies
4 samples have 3 copies, an additional copy to the chr5 duplication onto chr13
Breakpoints
PV0194-C
IGS-CBD-044
IGS-CBD-093
IGS−CBD−122
Code
cat /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200.bed | egrep "heptatricopeptide repeat-containing protein, putative" > PF3D7_0523200_regions.bed
cat /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200.bed | egrep "mitochondrial-processing peptidase subunit alpha" > PF3D7_0523100_regions.bedPV0194-C
Code
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/PV0194-C.sorted.bam --bed PF3D7_0523200_regions.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" > PV0194-C_PF3D7_0523200_regions_mates.bed
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/PV0194-C.sorted.bam --bed PF3D7_0523200_regions.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN | elucidator getOverlappingBedRegions --intersectWithBed STDIN --bed PV0194-C_PF3D7_0523200_regions_mates.bed | cut -f7 | sort | uniq -c
elucidator createBedRegionFromName --names Pf3D7_05_v3-947967-948435 > Pf3D7_05_v3-947967-948435.bed
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/PV0194-C.sorted.bam --bed Pf3D7_05_v3-947967-948435.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" > PV0194-C_Pf3D7_05_v3-947967-948435_mates.bed
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/PV0194-C.sorted.bam --bed Pf3D7_05_v3-947967-948435.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN | elucidator getOverlappingBedRegions --intersectWithBed STDIN --bed PV0194-C_Pf3D7_05_v3-947967-948435_mates.bed | cut -f7 | sort | uniq -c
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/PV0194-C.sorted.bam --bed Pf3D7_05_v3-947967-948435.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN | elucidator getOverlappingBedRegions --intersectWithBed STDIN --bed PV0194-C_Pf3D7_05_v3-947967-948435_mates.bed | cut -f7 | sort | uniq | egrep f3D7_05_v3-969 | elucidator createBedRegionFromName --names STDIN | bedtools sort | bedtools merge -d 10 | elucidator bed3ToBed6 --bed STDIN | egrep Pf3D7_05_v3-969374-969840 > Pf3D7_05_v3-969374-969840.bed
cat Pf3D7_05_v3-947967-948435.bed Pf3D7_05_v3-969374-969840.bed > combined_Pf3D7_05_v3-947967-948435_Pf3D7_05_v3-969374-969840.bed
cat Pf3D7_05_v3-947967-948435.bed Pf3D7_05_v3-969374-969840.bed | elucidator extendBedRegions --bed STDIN --left 25 --right 25 | sed -E 's/Pf3D7_05_v3-[0-9]+-[0-9]+/combined_Pf3D7_05_v3-947967-948435_Pf3D7_05_v3-969374-969840/g' > combined_Pf3D7_05_v3-947967-948435_Pf3D7_05_v3-969374-969840_l25_r25.bed
PathWeaver BamExtractPathwaysFromRegion --bed combined_Pf3D7_05_v3-947967-948435_Pf3D7_05_v3-969374-969840_l25_r25.bed --genomeDir /tank/data/genomes/plasmodium/genomes/pf/genomes/ --primaryGenome Pf3D7 --bam ../bams/PV0194-C.sorted.bam --dout combined_Pf3D7_05_v3-947967-948435_Pf3D7_05_v3-969374-969840_l25_r25/PV0194-C_combined_Pf3D7_05_v3-947967-948435_Pf3D7_05_v3-969374-969840_l25_r25 --overWriteDir --numThreads 4 --keepTemporaryFiles --qualCheck 5 --keepImproperMates
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/PV0194-C.sorted.bam --bed combined_Pf3D7_05_v3-947967-948435_Pf3D7_05_v3-969374-969840.bed > pairsInfo_combined_Pf3D7_05_v3-947967-948435_Pf3D7_05_v3-969374-969840_PV0194-C.tsv970043
IGS-CBD-044
946684 Ax4Tx4Ax2Gx1Ax22 (33 total), 964504 Ax36
Code
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-044.sorted.bam --bed PF3D7_0523100_regions.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" > IGS-CBD-044_PF3D7_0523100_regions_mates.bed
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-044.sorted.bam --bed PF3D7_0523100_regions.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN | elucidator getOverlappingBedRegions --intersectWithBed STDIN --bed IGS-CBD-044_PF3D7_0523100_regions_mates.bed | cut -f7 | sort | uniq -c
elucidator createBedRegionFromName --names Pf3D7_05_v3-946684-946894 > Pf3D7_05_v3-946684-946894.bed
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-044.sorted.bam --bed Pf3D7_05_v3-946684-946894.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" > IGS-CBD-044_Pf3D7_05_v3-946684-946894_mates.bed
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-044.sorted.bam --bed Pf3D7_05_v3-946684-946894.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN | elucidator getOverlappingBedRegions --intersectWithBed STDIN --bed IGS-CBD-044_Pf3D7_05_v3-946684-946894_mates.bed | cut -f7 | sort | uniq -c
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-044.sorted.bam --bed Pf3D7_05_v3-946684-946894.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN | elucidator getOverlappingBedRegions --intersectWithBed STDIN --bed IGS-CBD-044_Pf3D7_05_v3-946684-946894_mates.bed | cut -f7 | sort | uniq | egrep Pf3D7_05_v3-964292-964571 | elucidator createBedRegionFromName --names STDIN | bedtools sort | bedtools merge -d 10 | elucidator bed3ToBed6 --bed STDIN | egrep Pf3D7_05_v3-964292-964571 > Pf3D7_05_v3-964292-964571.bed
cat Pf3D7_05_v3-946684-946894.bed Pf3D7_05_v3-964292-964571.bed > combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964292-964571.bed
cat Pf3D7_05_v3-946684-946894.bed Pf3D7_05_v3-964292-964571.bed | elucidator extendBedRegions --bed STDIN --left 25 --right 25 | sed -E 's/Pf3D7_05_v3-[0-9]+-[0-9]+/combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964292-964571/g' > combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964292-964571_l25_r25.bed
PathWeaver BamExtractPathwaysFromRegion --bed combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964292-964571_l25_r25.bed --genomeDir /tank/data/genomes/plasmodium/genomes/pf/genomes/ --primaryGenome Pf3D7 --bam ../bams/IGS-CBD-044.sorted.bam --dout combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964292-964571_l25_r25/IGS-CBD-044_combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964292-964571_l25_r25 --overWriteDir --numThreads 4 --keepTemporaryFiles --qualCheck 5 --keepImproperMates
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-044.sorted.bam --bed combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964292-964571.bed > pairsInfo_combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964292-964571_IGS-CBD-044.tsvIGS-CBD-093
946684 Ax4Tx4Ax2Gx1Ax22 (33 total), 964504 Ax36
Code
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-093.sorted.bam --bed PF3D7_0523100_regions.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" > IGS-CBD-093_PF3D7_0523100_regions_mates.bed
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-093.sorted.bam --bed PF3D7_0523100_regions.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN | elucidator getOverlappingBedRegions --intersectWithBed STDIN --bed IGS-CBD-093_PF3D7_0523100_regions_mates.bed | cut -f7 | sort | uniq -c
elucidator createBedRegionFromName --names Pf3D7_05_v3-946684-946894 > Pf3D7_05_v3-946684-946894.bed
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-093.sorted.bam --bed Pf3D7_05_v3-946684-946894.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" > IGS-CBD-093_Pf3D7_05_v3-946684-946894_mates.bed
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-093.sorted.bam --bed Pf3D7_05_v3-946684-946894.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN | elucidator getOverlappingBedRegions --intersectWithBed STDIN --bed IGS-CBD-093_Pf3D7_05_v3-946684-946894_mates.bed | cut -f7 | sort | uniq -c
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-093.sorted.bam --bed Pf3D7_05_v3-946684-946894.bed | cut -f16-21 | tail -n +2 | egrep -v "\*" | bedtools sort | bedtools merge | elucidator bed3ToBed6 --bed STDIN | elucidator getOverlappingBedRegions --intersectWithBed STDIN --bed IGS-CBD-093_Pf3D7_05_v3-946684-946894_mates.bed | cut -f7 | sort | uniq | egrep Pf3D7_05_v3-964277-964567 | elucidator createBedRegionFromName --names STDIN | bedtools sort | bedtools merge -d 10 | elucidator bed3ToBed6 --bed STDIN | egrep Pf3D7_05_v3-964277-964567 > Pf3D7_05_v3-964277-964567.bed
cat Pf3D7_05_v3-946684-946894.bed Pf3D7_05_v3-964277-964567.bed > combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964277-964567.bed
cat Pf3D7_05_v3-946684-946894.bed Pf3D7_05_v3-964277-964567.bed | elucidator extendBedRegions --bed STDIN --left 25 --right 25 | sed -E 's/Pf3D7_05_v3-[0-9]+-[0-9]+/combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964277-964567/g' > combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964277-964567_l25_r25.bed
PathWeaver BamExtractPathwaysFromRegion --bed combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964277-964567_l25_r25.bed --genomeDir /tank/data/genomes/plasmodium/genomes/pf/genomes/ --primaryGenome Pf3D7 --bam ../bams/IGS-CBD-093.sorted.bam --dout combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964277-964567_l25_r25/IGS-CBD-093_combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964277-964567_l25_r25 --overWriteDir --numThreads 4 --keepTemporaryFiles --qualCheck 5 --keepImproperMates
elucidator getMateMapLocationForRegion --bam /tank/data/plasmodium/falciparum/pfdata/bams/IGS-CBD-093.sorted.bam --bed combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964277-964567.bed | egrep -v "\*" > pairsInfo_combined_Pf3D7_05_v3-946684-946894_Pf3D7_05_v3-964277-964567_IGS-CBD-093.tsvCopy number variation
Code
mdrCopyNumbers_byRawSample = tibble(
sample = c("PV0194-C",
"IGS-CBD-093",
"IGS-CBD-044",
"IGS-CBD-122",
"PH0529-C",
"PH0416-C",
"PH0397-C",
"PH0413-C",
"PH0229-C",
"PH0534-C",
"PH0274-C",
"PH0480-C",
"PH0959-Cx",
"IVC11-BB-0084",
"PH0544-C",
"PD1430-C",
"PH0681-C",
"PH0697-C",
"PH0894-C",
"PH0817-C",
"IGS-CBD-015",
"IGS-CBD-020",
"IGS-CBD-060",
"PH0143-CW",
"PH0278-CW",
"PH0308-CW",
"PH0474-CW",
"PH0457-CW",
"PH0467-CW",
"PC0019-C",
"PH0149-CW",
"PH0153-CW",
"PH0395-C",
"PV0035-C",
"PH0387-C",
"QE0455-C",
"PV0142-C",
"PV0112-C",
"PV0098-C",
"PF0584-C",
"PH0156-C",
"PV0257-C",
"PH0412-C",
"PV0274-C",
"PD1299-C",
"PH1310-C",
"IGS-CBD-036",
"PH1616-C",
"PT0023-CW",
"PV0097-C"
),
`pfmdr11 Copies` = c(
"3",
"3",
"3",
"3",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"2",
"1",
"1",
"1",
"1",
"1",
"1",
"1",
"1",
"1",
"1",
"1",
"1",
"2",
"1",
"1",
"1",
"1",
"2",
"1",
"1",
"1"
)
)
mdrCopyNumbers = mdrCopyNumbers_byRawSample %>%
left_join(allMetaDeletionCalls %>%
select(sample, BiologicalSample)) %>%
mutate(sample = BiologicalSample) %>%
select(-BiologicalSample)Breakpoints
Likely breakpoints
969840
PV0194-C 947967 Ax19, 970043 Ax18
Pf3D7_05_v3 947967 947996 Pf3D7_05_v3-947967-947996 29 +
Pf3D7_05_v3 947957 947996 Pf3D7_05_v3-947957-947996 39 +
IGS-CBD-044 946684 Ax4Tx4Ax2Gx1Ax22 (33 total), 964504 Ax36
IGS-CBD-093 946684 Ax4Tx4Ax2Gx1Ax22 (33 total), 964504 Ax36
IGS−CBD−122 946684 Ax4Tx4Ax2Gx1Ax22 (33 total), 964504 Ax36
Plotting coverage around pfmdr1 duplication
Downloads
Code
cov = readr::read_tsv("../../meta/allCov_summaryStats.tab.txt.gz")
allMetaDeletionCalls_Hrp3pattern2_samples = readr::read_tsv("/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt", col_names = "sample")
allReports = readr::read_tsv("stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200/popClustering//reports/allBasicInfo.tab.txt.gz") %>%
filter(start >= 944389) %>%
filter(sample %in% allMetaDeletionCalls_Hrp3pattern2_samples$sample) %>%
# filter(sample %in% allMetaDeletionCalls$sample) %>%
mutate(inGene = !is.na(extraField0) ) %>%
left_join(cov) %>%
mutate(medianCov = median(perBaseCoverage),
meanCov = mean(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = ifelse(inGene, perBaseCoverage/medianPerBaseCov_inGenes, perBaseCoverage/medianPerBaseCov_notInGenes)) %>%
mutate(perBaseCoverageNormRounded = ifelse(perBaseCoverageNorm >0.10 & perBaseCoverageNorm <= 1, 1, perBaseCoverageNorm)) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNormRounded)) %>%
filter(sample %!in% realmccoilCoiCalls_poly$sample)
allReports_sp = allReports %>%
select(sample, name, perBaseCoverageNormRounded) %>%
spread(name, perBaseCoverageNormRounded)
allReports_sp_mat = as.matrix(allReports_sp[,2:ncol(allReports_sp)])
rownames(allReports_sp_mat) = allReports_sp$sample
allReports_sp_mat[allReports_sp_mat >3] = 3
annotationTextSize = 20
allRegionsOfChromosome5Region = allReports %>%
filter(sample == .$sample[1])
allRegionsOfChromosome5Region_filt = allRegionsOfChromosome5Region %>%
filter(start >= 952574, end <= 979203)
allRegionsOfChromosome5Region_filt_sel = allRegionsOfChromosome5Region_filt %>%
select(extraField0) %>%
unique() %>%
filter(!is.na(extraField0)) %>%
mutate(ID = gsub(";.*", "", gsub(".*ID=", "", extraField0))) %>%
mutate(description = gsub("\\]", "", gsub(".*description=", "", extraField0)))
create_dt(allRegionsOfChromosome5Region_filt_sel)Code
topAnno = allReports %>%
filter(sample == .$sample[1]) %>%
select(name, extraField0) %>%
rename(`Gene Description` = extraField0) %>%
mutate(`Gene Description` = gsub(".*description=", "", `Gene Description`))%>%
mutate(`Gene Description` = gsub("\\].*", "", `Gene Description`)) %>%
mutate(`Gene Description` = ifelse(grepl("PHIST", `Gene Description`), gsub("\\).*", "", gsub(".*PHIST", "PHIST", `Gene Description`)), `Gene Description`))
transitionPoint = allReports %>%
filter(sample == .$sample[1]) %>%
mutate(diffFromTransition = abs(979203 - start)) %>%
mutate(`Transition Point` = ifelse(diffFromTransition <=100, TRUE, NA)) %>%
select(name, `Transition Point`)
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_sampleRegion_count = stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_countTares_sum_filt %>%
group_by(sample) %>%
group_by(region) %>%
count(name = "TARE1 Present Chr5 Count")
topAnno = topAnno %>%
left_join(
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_sampleRegion_count %>%
rename(name = region)
) %>%
left_join(transitionPoint) %>%
separate(name, into = c("chrom", "start", "end", "strand"), sep = "-", convert = T, remove = F) %>%
mutate(`Genomic Region` = ifelse(start >=952484 & end <= 979203, "Chr5 Duplication\nOnto Chr13", NA)) %>%
select(-chrom, -start, -end, -strand, -`TARE1 Present Chr5 Count`, -`Transition Point`)
topAnnoDf = topAnno %>%
mutate(`Gene Description` = gsub("multidrug resistance protein 1", "multidrug resistance protein 1(pfmdr1)", `Gene Description`)) %>%
select(-name) %>%
as.data.frame()
topAnnoColors = createColorListFromDf(topAnnoDf)
topAnnoColors[["Genomic Region"]] = c("Chr11/13 Duplicated Region" = "#0072B2", "Subtelomere" = "#E69F00", "Chr5 Duplication\nOnto Chr13" = "#EF0096")
topAnnoColors_previousGeneDescriptionsColors = topAnnoColors$`Gene Description`
metaReSelected = allMetaDeletionCalls[match(rownames(allReports_sp_mat), allMetaDeletionCalls$sample), ]
# rowAnno = tibble(metaReSelected[,c("sample","country", "secondaryRegion")]) %>%
# rename(continent = secondaryRegion) %>%
# mutate(`TARE1 On Chr13` = sample %in% hrp3Pat2_samplesWithTare1$sample,
# `Transposition With Chr5` = sample %in% samplesWithTransitionToChr5$sample,
# `TARE1 On Chr5` = sample %in% stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_sampleRegion$sample)
rowAnno = tibble(metaReSelected[,c("sample","country", "secondaryRegion")]) %>%
rename(continent = secondaryRegion) %>%
mutate(`TARE1 On Chr13` = sample %in% hrp3Pat2_samplesWithTare1$sample,
`Transposition With Chr5` = sample %in% samplesWithTransitionToChr5$sample,
`TARE1 On Chr5` = sample %in% stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_countTares_sum_filt$sample) %>%
mutate(`TARE1 On Chr13` = ifelse(`TARE1 On Chr13`, `TARE1 On Chr13`, NA))%>%
mutate(`Transposition With Chr5` = ifelse(`Transposition With Chr5`, `Transposition With Chr5`, NA))%>%
mutate(`TARE1 On Chr5` = ifelse(`TARE1 On Chr5`, `TARE1 On Chr5`, NA)) %>%
left_join(mdrCopyNumbers_byRawSample)
write_tsv(rowAnno,
file = "stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_sampleRegion_hrp3_pat2_sample_deletion_meta.tsv")
rowAnnoDf = rowAnno %>%
select(-sample) %>%
select(`pfmdr11 Copies`, country, continent, `TARE1 On Chr13`, `Transposition With Chr5`, `TARE1 On Chr5`) %>%
as.data.frame()
rowAnnoColors = createColorListFromDf(rowAnnoDf)
rowAnnoColors[["continent"]] = c("AFRICA" = "#E69F00",
"ASIA" = "#F0E442",
"OCEANIA" = "#F748A5",
"S_AMERICA" = "#0072B2")
rowAnnoColors[["TARE1 On Chr5"]] = c("TRUE" = "#008607")
rowAnnoColors[["Transposition With Chr5"]] = c("TRUE" = "#008607")
rowAnnoColors[["TARE1 On Chr13"]] = c("TRUE" = "#008607")
sideAnno = rowAnnotation(
df = rowAnnoDf,
col = rowAnnoColors,
gp = gpar(col = "grey10"),
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
na_col = c("#99999900"),
gap = unit(0.1, "cm"),
show_legend = c("country"= T, "continent"= T, "TARE1 On Chr13" = F, "Transposition With Chr5" = F, "TARE1 On Chr5" = F)
)
topAnnoHM = HeatmapAnnotation(
df = topAnnoDf,
col = topAnnoColors,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
na_col = c("#99999900"),
gap = unit(0.1, "cm")
)
library(circlize)
col_fun = colorRamp2(c(0, max(allReports_sp_mat)/2, max(allReports_sp_mat)), c(heat.colors(3)))
allReports_sp_mat_nolab = allReports_sp_mat
colnames(allReports_sp_mat_nolab) = NULL
# rownames(allReports_sp_mat_nolab) = NULL
allReports_sp_mat_hm = Heatmap(
allReports_sp_mat_nolab,
cluster_columns = F,
col = col_fun,
name = "coverage",
top_annotation = topAnnoHM,
right_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
at = c(0, 0.5, 1, 1.5, 2, 2.5, 3),
labels = c("0", "0.5x", "1.0x", "1.5x", "2x", "2.5x", ">=3x")
),
cell_fun = function(j, i, x, y, width, height, fill) {
grid.rect(x = x, y = y, width = width, height = height *.9,
gp = gpar(fill = fill, col = NA))
},
rect_gp = gpar(type = "none"),
column_title = "Chr05[944kb:988kb]",
column_title_gp = gpar(fontsize = 20, fontface = "italic")
)
#
allReports_sp_mat_hm_noCluster = Heatmap(
allReports_sp_mat_nolab,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnnoHM,
right_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
at = c(0, 0.5, 1, 1.5, 2, 2.5, 3),
labels = c("0", "0.5x", "1.0x", "1.5x", "2x", "2.5x", ">=3x")
),
cell_fun = function(j, i, x, y, width, height, fill) {
grid.rect(x = x, y = y, width = width, height = height *.9,
gp = gpar(fill = fill, col = NA))
},
rect_gp = gpar(type = "none"),
column_title = "Chr05[944kb:988kb]",
column_title_gp = gpar(fontsize = 20, fontface = "italic")
)pfmdr1 final plot
Code
draw(allReports_sp_mat_hm, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom")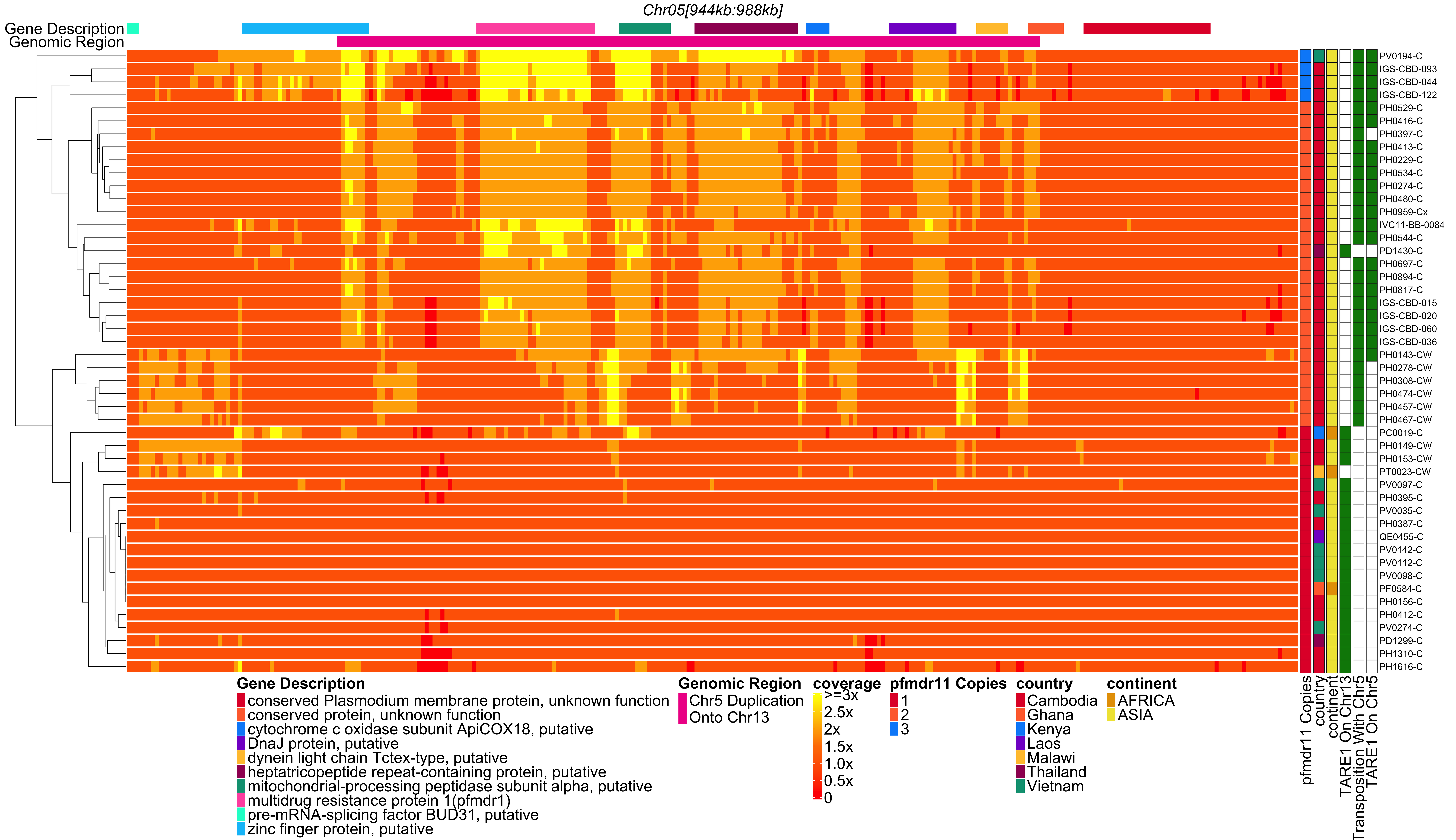
Code
quartz_off_screen
2 Code
Code
fillterDf = expand_grid(sample = rownames(allReports_sp_mat),
region = colnames(allReports_sp_mat)) %>%
mutate(marker = 0)
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded = stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_countTares %>%
select(sample, region) %>%
mutate(marker = 1) %>%
bind_rows(fillterDf) %>%
group_by(sample, region) %>%
summarise(marker = sum(marker))
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp = stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded %>%
spread(region, marker)
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp_mat = as.matrix(stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp[,2:ncol(stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp)])
rownames(stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp_mat) = stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp$sample
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp_mat = stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp_mat[match(
rownames(allReports_sp_mat), rownames(stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp_mat)
), ]
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp_mat_nolab = stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp_mat
colnames(stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp_mat_nolab) = NULL
#rownames(stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp_mat_nolab) = NULL
allReports_sp_mat_hm_tare1 = Heatmap(
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_expanded_sp_mat_nolab,
cluster_columns = F,
cluster_rows = F,
col = colorRamp2(c(0, 1), c("#FFFFFF00","green")),
name = "coverage",
top_annotation = topAnnoHM,
right_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
at = c(0, 0.5, 1, 1.5, 2, 2.5, 3),
labels = c("0", "0.5x", "1.0x", "1.5x", "2x", "2.5x", ">=3x")
),
cell_fun = function(j, i, x, y, width, height, fill) {
grid.rect(x = x, y = y, width = width, height = height *.9,
gp = gpar(fill = fill, col = NA))
},
rect_gp = gpar(type = "none")
)Code
pdf("stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_withTare.pdf", width = 32, height = 25, useDingbats = F)
draw(allReports_sp_mat_hm_noCluster, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom")
draw(allReports_sp_mat_hm_tare1, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom")
dev.off()quartz_off_screen
2 Code
elucidator createWindowsInRegions --bed stableWindows_05_regionOfInterestNarrow.bed --step 50 --windowSize 100 --minLen 50 | elucidator bedGetIntersectingGenesInGff --gff /tank/data/genomes/plasmodium/genomes/pf/info/gff/Pf3D7.gff --extraAttributes description --bed STDIN --overWrite --out stableWindows_05_regionOfInterestNarrow_step50_windowSize100.bed
elucidator createWindowsInRegions --bed stableWindows_05_regionOfInterestNarrow.bed --step 150 --windowSize 200 --minLen 50 | elucidator bedGetIntersectingGenesInGff --gff /tank/data/genomes/plasmodium/genomes/pf/info/gff/Pf3D7.gff --extraAttributes description --bed STDIN --overWrite --out stableWindows_05_regionOfInterestNarrow_step150_windowSize200.bed
rsync -raPh stableWindows_05_regionOfInterestNarrow.bed hathawan@calderon.barrel-of-knowledge.info:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22
rsync -raPh stableWindows_05_regionOfInterestNarrow_step50_windowSize100.bed hathawan@calderon.barrel-of-knowledge.info:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22
rsync -raPh stableWindows_05_regionOfInterestNarrow_step150_windowSize200.bed hathawan@calderon.barrel-of-knowledge.info:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22Code
nohup elucidator runMultipleCommands --cmdFile runRegionBamTrimCmds.txt --additionalFields "{DIRNAME}:stableWindows_05_regionOfInterestNarrow;{SAMPLE}:allSamples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/stableWindows_05_regionOfInterestNarrow.bed\"" --numThreads 11 &
cd stableWindows_05_regionOfInterestNarrow
PathWeaver runProcessClustersOnRecon --overWriteDir --dout popClusteringSWGAErrors --pat _stableWindows_05_regionOfInterestNarrow --numThreads 5 --groupingsFile /tank/data/plasmodium/falciparum/pfdata/metadata/meta.tab.txt
rm -f runSimpleCallVariantCmds.txt && mkdir -p reports/varCalls && mkdir -p reports/alnCaches && for x in Pf*; do echo elucidator callVariantsAgainstRefGenome --fastagz ${x}/population/popSeqsWithMetaWtihSampleName.fasta.gz --genome /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.2bit --ignoreSubFields site:LabCross,site:LabControl,site:LabContaminated --overWriteDir --lowVariantCutOff 0.00 --occurrenceCutOff 2 --dout reports/varCalls/vars_${x} --alnInfoDir reports/alnCaches/alnCache_${x} --gff /tank/data/genomes/plasmodium/genomes/pf/info/gff/Pf3D7.gff --knownAminoAcidChangesFnp /tank/data/genomes/plasmodium/genomes/pf/info/pf_drug_resistant_aaPositions.tsv --getPairwiseComps --identifier ${x} --metaFieldsToCalcPopDiffs region,country,secondaryRegion >> runSimpleCallVariantCmds.txt; done;
nohup elucidator runMultipleCommands --cmdFile runSimpleCallVariantCmds.txt --numThreads 10 --raw &
rm -fr runSubSegmentCmds.txt && mkdir -p reports/subSegments && for x in Pf3D7_05_v3-9*; do echo elucidator createSharedSubSegmentsFromRefSeqs --genome /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta --refBedFnp /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/stableWindows_05_regionOfInterestNarrow.bed --refBedID ${x} --fastagz ${x}/population/popSeqsWithMetaWtihSampleName.fasta.gz --dout reports/subSegments/${x}_subSegments_corrected2 --correctionOccurenceCutOff 2 --lowFreqCutOff 2 --overWriteDir --lenFilter --kSimFilter >> runSubSegmentCmds.txt; done;
nohup elucidator runMultipleCommands --cmdFile runSubSegmentCmds.txt --numThreads 10 --raw &
cd reports/subSegments
elucidator rBind --contains 0_ref_variable_expanded_genomic.bed --delim tab --recursive | elucidator bedGetIntersectingGenesInGff --gff /tank/data/genomes/plasmodium/genomes/pf/info/gff/Pf3D7.gff --extraAttributes description --bed STDIN --overWrite --out stableWindows_05_regionOfInterestNarrow_allRefVariableRegions.bedCode
nohup elucidator runMultipleCommands --cmdFile runRegionBamTrimCmds.txt --additionalFields "{DIRNAME}:stableWindows_05_regionOfInterestNarrow_allRefVariableRegions;{SAMPLE}:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/stableWindows_05_regionOfInterestNarrow_allRefVariableRegions.bed\";{NCPUS}:4" --replaceFields --numThreads 11 &
nohup elucidator runMultipleCommands --cmdFile runRegionBamTrimCmds.txt --additionalFields "{DIRNAME}:stableWindows_05_regionOfInterestNarrow_allRefVariableRegions;{SAMPLE}:/tank/data/plasmodium/falciparum/pfdata/metadata/mixtureInfo/monoclonalControlWGSSamples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/stableWindows_05_regionOfInterestNarrow_allRefVariableRegions.bed\";{NCPUS}:4" --replaceFields --numThreads 11 &
cd stableWindows_05_regionOfInterestNarrow_allRefVariableRegions
PathWeaver runProcessClustersOnRecon --overWriteDir --dout popClustering --pat _stableWindows_05_regionOfInterestNarrow_allRefVariableRegions --numThreads 5 --groupingsFile /tank/data/plasmodium/falciparum/pfdata/metadata/meta.tab.txthaplotypes
stableWindows_05_regionOfInterestNarrow variable windows
Code
cov = readr::read_tsv("../../meta/allCov_summaryStats.tab.txt.gz")
allMetaDeletionCalls_Hrp3pattern2_samples = readr::read_tsv("/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt", col_names = "sample")
allReports = readr::read_tsv("stableWindows_05_regionOfInterestNarrow_allRefVariableRegions/popClustering/reports/allBasicInfo.tab.txt.gz") %>%
filter(sample %in% allMetaDeletionCalls_Hrp3pattern2_samples$sample) %>%
mutate(inGene = !is.na(extraField0) ) %>%
left_join(cov) %>%
mutate(medianCov = median(perBaseCoverage),
meanCov = mean(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = ifelse(inGene, perBaseCoverage/medianPerBaseCov_inGenes, perBaseCoverage/medianPerBaseCov_notInGenes)) %>%
mutate(perBaseCoverageNormRounded = ifelse(perBaseCoverageNorm >0.10 & perBaseCoverageNorm <= 1, 1, perBaseCoverageNorm)) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNormRounded)) %>%
filter(sample %!in% realmccoilCoiCalls_poly$sample)
allReports_sp = allReports %>%
select(sample, name, perBaseCoverageNormRounded) %>%
spread(name, perBaseCoverageNormRounded)
allReports_sp_mat = as.matrix(allReports_sp[,2:ncol(allReports_sp)])
rownames(allReports_sp_mat) = allReports_sp$sample
allReports_sp_mat[allReports_sp_mat >2] = 2
annotationTextSize = 20
allRegionsOfChromosome5Region = allReports %>%
filter(sample == .$sample[1])
allRegionsOfChromosome5Region_filt = allRegionsOfChromosome5Region %>%
filter(start >= 952574, end <= 979203)
allRegionsOfChromosome5Region_filt_sel = allRegionsOfChromosome5Region_filt %>%
select(extraField0) %>%
unique() %>%
filter(!is.na(extraField0)) %>%
mutate(ID = gsub(";.*", "", gsub(".*ID=", "", extraField0))) %>%
mutate(description = gsub("\\]", "", gsub(".*description=", "", extraField0)))
create_dt(allRegionsOfChromosome5Region_filt_sel)Code
topAnno = allReports %>%
filter(sample == .$sample[1]) %>%
select(name, extraField0) %>%
mutate(name = gsub("\\.", "-", name)) %>%
rename(`Gene Description` = extraField0) %>%
mutate(`Gene Description` = gsub(".*description=", "", `Gene Description`))%>%
mutate(`Gene Description` = gsub("\\].*", "", `Gene Description`)) %>%
mutate(`Gene Description` = ifelse(grepl("PHIST", `Gene Description`), gsub("\\).*", "", gsub(".*PHIST", "PHIST", `Gene Description`)), `Gene Description`))
transitionPoint = allReports %>%
filter(sample == .$sample[1]) %>%
mutate(diffFromTransition = abs(979203 - start)) %>%
mutate(`Transition Point` = ifelse(diffFromTransition <=100, TRUE, NA)) %>%
select(name, `Transition Point`)
topAnno = topAnno
topAnnoDf = topAnno %>%
select(-name) %>%
as.data.frame()
topAnnoColors = createColorListFromDf(topAnnoDf)
metaReSelected = allMetaDeletionCalls[match(rownames(allReports_sp_mat), allMetaDeletionCalls$sample), ]
rowAnno = tibble(metaReSelected[,c("sample","country", "secondaryRegion", "collection_year")]) %>%
rename(continent = secondaryRegion) %>%
mutate(`TARE1 On Chr13` = sample %in% hrp3Pat2_samplesWithTare1$sample,
`Transposition With Chr5` = sample %in% samplesWithTransitionToChr5$sample,
`TARE1 On Chr5` = sample %in% stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_countTares_sum_filt$sample)
rowAnnoDf = rowAnno %>%
select(-sample) %>%
as.data.frame()
rowAnnoColors = createColorListFromDf(rowAnnoDf)
rowAnnoColors[["continent"]] = c("AFRICA" = "#E69F00",
"ASIA" = "#F0E442",
"OCEANIA" = "#F748A5",
"S_AMERICA" = "#0072B2")
sideAnno = rowAnnotation(
df = rowAnnoDf,
col = rowAnnoColors,
gp = gpar(col = "grey10"),
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
gap = unit(0.1, "cm")
)
topAnnoHM = HeatmapAnnotation(
df = topAnnoDf,
col = topAnnoColors,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left"
)
library(circlize)
col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3)))
allReports_sp_mat_nolab = allReports_sp_mat
colnames(allReports_sp_mat_nolab) = NULL
allReports_sp_mat_hm = Heatmap(
allReports_sp_mat_nolab,
cluster_columns = F,
col = col_fun,
name = "coverage",
top_annotation = topAnnoHM,
right_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
cell_fun = function(j, i, x, y, width, height, fill) {
grid.rect(x = x, y = y, width = width, height = height *.9,
gp = gpar(fill = fill, col = NA))
},
rect_gp = gpar(type = "none")
)Code
draw(allReports_sp_mat_hm, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom")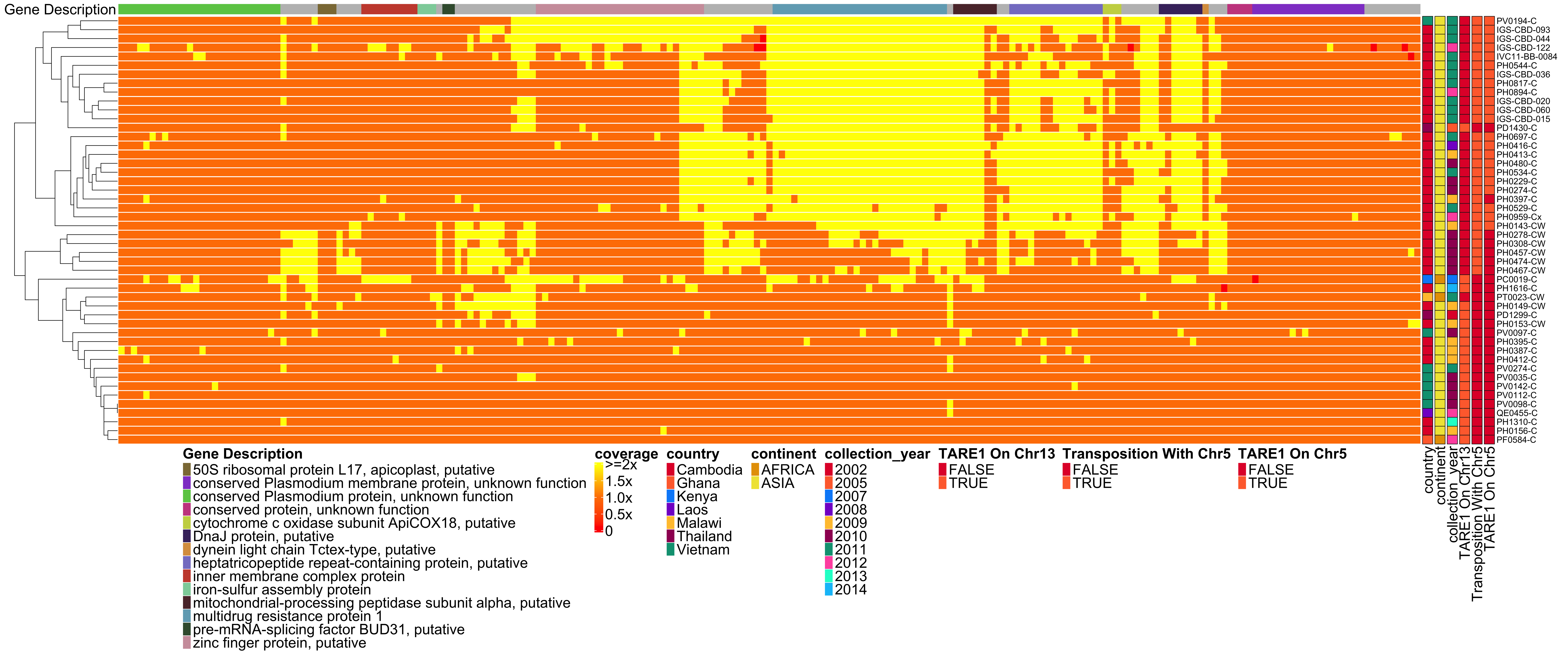
Plotting the haplotype variation around pfmdr1 duplication
Downloads
Code
allMetaSamples_filt = allMetaSamples %>%
filter(sample %in% allMetaDeletionCalls_Hrp3pattern2_samples$sample) %>%
filter(sample %!in% realmccoilCoiCalls_poly$sample)
allMetaSamplesByBio_filt = allMetaSamplesByBio %>%
filter(site == "LabIsolate")
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering = readr::read_tsv("stableWindows_05_regionOfInterestNarrow_allRefVariableRegions/popClustering/reports/allSelectedClustersInfo.tab.txt.gz") %>%
#filter(s_Sample %in% c(allMetaSamplesByBio_filt$sample, allMetaSamples_filt$BiologicalSample))%>%
filter(s_Sample %in% c(allMetaSamples_filt$BiologicalSample))
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering = stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering %>%
separate(p_name, remove = F, convert = T, sep = "-", into = c("chrom", "start", "end", "strand"))
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea = stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering %>%
filter(start >= 952574,
end <= 979381)
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep = HaplotypeRainbows::prepForRainbow(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea, minPopSize = 2)%>%
mutate(popid = ifelse(maxPopid == 1, -1, popid))
newRowAnno = tibble(sample = unique(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea$s_Sample))
newRowAnno = newRowAnno %>%
left_join(allMetaSamplesByBio) %>%
left_join(rowAnno) %>%
mutate(continent = secondaryRegion)
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp = stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep %>%
group_by(p_name) %>%
mutate(sampleCount = length(unique(s_Sample))) %>%
group_by() %>%
filter(sampleCount >= 0.90*max(sampleCount)) %>%
group_by(s_Sample, p_name) %>%
#filter(c_AveragedFrac == max(c_AveragedFrac)) %>%
mutate(marker = 1) %>%
group_by() %>%
select(h_popUID, marker, s_Sample) %>%
spread(h_popUID, marker, fill = 0)
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp_mat = as.matrix(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp[,2:ncol(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp)])
rownames(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp_mat) = stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp$s_Sample
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp_dist = dist(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp_mat)
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp_dist_hclust = hclust(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp_dist)
# stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_levels = c("PV0097-C","PC0019-C","PH0395-C","PH0412-C","PH0387-C","PH0156-C","PH0149-C","PH0153-C","PV0142-C","PV0098-C","PV0112-C","PD1430-C","PD1299-C","PH1616-C", "PT0023-C","PV0035-C","PH1310-C","PF0584-C","PH0467-C","PH0474-C","PV0257-C","QE0455-C","PV0274-C","PH0681-C","PH0457-C","PH0229-C","PH0480-C","PH0274-C","PH0308-CW","IGS-CBD-015","PH0959-Cx","IGS-CBD-060","PH0894-C","IGS-CBD-036","PH0697-C","PH0534-C","PH0416-C","PH0413-C","PH0143-C","PH0397-C","IVC11-BB-0084","PH0817-C","PH0544-C","PH0529-C","PH0278-CW","IGS-CBD-020","IGS-CBD-122","IGS-CBD-093","IGS-CBD-044","PV0194-C"
# )
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_levels = c("PV0097-C","PC0019-C","PH0395-C","PH0412-C","PH0387-C","PH0156-C","PH0149-C","PH0153-C","PV0142-C","PV0098-C","PV0112-C","PD1430-C","PD1299-C","PH1616-C", "PT0023-C","PV0035-C","PH1310-C","PF0584-C","PH0467-C","PH0474-C","QE0455-C","PV0274-C","PH0457-C","PH0229-C","PH0480-C","PH0274-C","PH0308-CW","IGS-CBD-015","PH0959-Cx","IGS-CBD-060","PH0894-C","IGS-CBD-036","PH0697-C","PH0534-C","PH0416-C","PH0413-C","PH0143-C","PH0397-C","IVC11-BB-0084","PH0817-C","PH0544-C","PH0529-C","PH0278-CW","IGS-CBD-020","IGS-CBD-122","IGS-CBD-093","IGS-CBD-044","PV0194-C"
)
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep = stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep %>%
mutate(
s_Sample = factor(as.character(s_Sample),
levels = stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_levels,
# levels = rownames(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp_mat)[stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_sp_dist_hclust$order]
)
)
# newRowAnno_reOrdered = newRowAnno[match(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_levels, newRowAnno$sample, ),]
newRowAnno_reOrdered = newRowAnno[match(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_levels, newRowAnno$sample, ),] %>%
mutate(sample = factor(as.character(sample), levels = levels(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$s_Sample)))
newRowAnno_reOrdered = newRowAnno_reOrdered %>%
mutate(`TARE1 On Chr13` = ifelse(is.na(`TARE1 On Chr13`), F, `TARE1 On Chr13`)) %>%
mutate(`Transposition With Chr5` = ifelse(is.na(`Transposition With Chr5`), F, `Transposition With Chr5`)) %>%
mutate(`TARE1 On Chr5` = ifelse(is.na(`TARE1 On Chr5`), F, `TARE1 On Chr5`)) %>%
left_join(mdrCopyNumbers)%>%
mutate(sample = factor(as.character(sample), levels = levels(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$s_Sample)))
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_plot = genRainbowHapPlotObj(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep, colorCol = popid) +
theme(axis.text.x = element_text(size=12, angle = -90, vjust = 0.5, hjust = 0)) +
scale_x_continuous(breaks = 1:length(levels(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$p_name)),
labels = levels(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$p_name))
allColors = c(); for(name in c("country", "Transposition With Chr5", "continent")){ allColors = c(allColors, rowAnnoColors[[name]])}
previousColors = unique(ggplot_build(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_plot)$data[[1]][["fill"]])
names(previousColors) = sort(unique(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$popid))
previousColors["-1"] = "grey0";
allColors = c(allColors, previousColors)
newrowAnnoColors = createColorListFromDf(newRowAnno_reOrdered)
newrowAnnoColors[["continent"]] = c("AFRICA" = "#E69F00",
"ASIA" = "#F0E442",
"OCEANIA" = "#F748A5",
"S_AMERICA" = "#0072B2")
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep = stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep %>%
mutate(popid= factor(popid))Code
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_withCountry_plot = genRainbowHapPlotObj(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep, colorCol = popid) +
theme(axis.text.x = element_text(size=12, angle = -90, vjust = 0.5, hjust = 0)) +
scale_x_continuous(breaks = c(-7.0 + 0.5, -5.5 + 0.5, -4 + 0.5, -2.5 + 0.5, -1 + 0.5, 1:length(levels(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$p_name))),
labels = c("", "", "", "", "", rep("", length(levels(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$p_name)))),
expand = c(0,0)) +
theme(axis.title.x = element_blank(),
panel.border = element_blank(),
axis.ticks = element_blank(),
axis.line.x = element_blank(),
axis.line.y = element_blank(),
legend.background = element_blank(),
legend.box.background = element_rect(colour = "black"),
legend.title = element_text(face = "bold")
#, text=element_text(size=21)
)+
theme(legend.text = element_text(size = 30),
axis.text.y = element_text(size = 30, color = "black"),
axis.text.x = element_text(size = 30, color = "black"),
plot.title = element_text(size = 30, color = "black"),
strip.text.y = element_text(size = 30, color = "black"),
legend.title = element_text(size = 30, face = "bold"),
legend.box="vertical", legend.margin=margin(),
legend.background = element_blank(),
legend.box.background = element_rect(colour = "black"))
#labels = c("Transposition With Chr5", "continent", "country", rep("", length(levels(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$p_name)))))
# labels = c("Transposition With Chr5", "continent", "country", levels(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$p_name)))
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_annotationLabelDf =tibble(
x = c(-7.0 + 0.5, -5.5 + 0.5, -4 + 0.5, -2.5 + 0.5, -1 + 0.5),
label = c("Transposition With Chr5", "continent", "country", "collection\nyear", "pfmdr11\nCopies")
)
topAnno_varInDup = topAnno %>%
filter(name %in% stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$p_name) %>%
mutate(name = factor(name, levels = levels(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$p_name))) %>%
mutate(`Gene Description` = gsub("multidrug resistance protein 1", "multidrug resistance protein 1(pfmdr1)", `Gene Description`))
stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_withCountry_plot = stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_withCountry_plot +
scale_fill_manual("Microhaplotype\nRank", values = haplotypeRankColors[sort(names(previousColors))], labels = names(haplotypeRankColors[sort(names(previousColors))]), breaks = names(haplotypeRankColors[sort(names(previousColors))])) +
guides(fill = guide_legend()) +
ggnewscale::new_scale_fill() +
geom_rect(aes(xmin= 0, xmax = -1,
ymin = as.numeric(sample) - 0.5,
ymax = as.numeric(sample) + 0.5,
fill = factor(`pfmdr11 Copies`)), color = "black", data = newRowAnno_reOrdered) +
scale_fill_manual("pfmdr11 Copies", values = newrowAnnoColors[["pfmdr11 Copies"]]) +
#guides(fill = guide_legend(ncol = 1)) +
ggnewscale::new_scale_fill() +
geom_rect(aes(xmin= -1.5, xmax = -2.5,
ymin = as.numeric(sample) - 0.5,
ymax = as.numeric(sample) + 0.5,
fill = factor(collection_year)), color = "black", data = newRowAnno_reOrdered) +
scale_fill_manual("collection_year", values = newrowAnnoColors[["collection_year"]],
guide = guide_legend(nrow = 3)) +
guides(fill = guide_legend(nrow = 3)) +
ggnewscale::new_scale_fill() +
geom_rect(aes(xmin= -3, xmax = -4,
ymin = as.numeric(sample) - 0.5,
ymax = as.numeric(sample) + 0.5,
fill = country), color = "black", data = newRowAnno_reOrdered) +
scale_fill_manual("country", values = newrowAnnoColors[["country"]]) +
#guides(fill = guide_legend(ncol = 1)) +
ggnewscale::new_scale_fill() +
geom_rect(aes(xmin= -4.5, xmax = -5.5,
ymin = as.numeric(sample) - 0.5,
ymax = as.numeric(sample) + 0.5,
fill = secondaryRegion), color = "black", data = newRowAnno_reOrdered) +
scale_fill_manual("Continent", values = newrowAnnoColors[["continent"]]) +
#guides(fill = guide_legend(ncol = 1)) +
ggnewscale::new_scale_fill() +
geom_rect(aes(xmin= -6, xmax = -7,
ymin = as.numeric(sample) - 0.5,
ymax = as.numeric(sample) + 0.5,
fill = `Transposition With Chr5`), color = "black", data = newRowAnno_reOrdered)+
scale_fill_manual("Transposition With Chr5", values = rowAnnoColors[["Transposition With Chr5"]]) +
guides(fill = guide_legend(nrow = 3)) +
ggnewscale::new_scale_fill() +
geom_rect(
aes(
xmin = as.numeric(name) - 0.5,
xmax = as.numeric(name) + 0.5,
ymax = 0,
ymin = -7,
fill = `Gene Description`
),
data = topAnno_varInDup,
color = "black"
) +
scale_fill_manual("Genes\nDescription",
# values = topAnnoColors[["Gene Description"]][names(topAnnoColors[["Gene Description"]]) %in% topAnno_varInDup$`Gene Description`],
values = topAnnoColors_previousGeneDescriptionsColors[names(topAnnoColors_previousGeneDescriptionsColors) %in% topAnno_varInDup$`Gene Description`],
guide = guide_legend(nrow = 6)) +
geom_text(
aes(x = 0.45, y = -7/2), label = "Genes\nDescription", hjust = 1, face = "bold", size = 7
) +
geom_text(
aes(x = x, y = 0, label = label),
angle = -90, hjust = 0,
size = 7,
data = stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_annotationLabelDf
) +
labs(x = "", y = "") +
scale_y_continuous(labels = levels(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$s_Sample),
breaks = 1:length(levels(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep$s_Sample)), expand = c(0,0)) +
transparentBackground + theme(legend.box.background = element_rect(colour = "black"))Code
print(stableWindows_05_regionOfInterestNarrow_allRefVariableRegions_popClustering_dupArea_prep_withCountry_plot)
100bp windows on chromosome 13
Downloads
Plot small windows on chromosome 13 with meta about patterns for 11/13-
Code
cov = readr::read_tsv("../../meta/allCov_summaryStats.tab.txt.gz")
allReports = readr::read_tsv("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step100_windowSize100_NoTrim_keep/reports/allBasicInfo.tab.txt.gz") %>%
mutate(inGene = !is.na(extraField0)) %>%
left_join(cov) %>%
mutate(medianCov = median(perBaseCoverage),
meanCov = mean(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = ifelse(inGene, perBaseCoverage/medianPerBaseCov_inGenes, perBaseCoverage/medianPerBaseCov_notInGenes)) %>%
mutate(perBaseCoverageNormRounded = ifelse(perBaseCoverageNorm >0.10 & perBaseCoverageNorm <= 1, 1, perBaseCoverageNorm)) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNormRounded)) %>%
filter(sample %!in% realmccoilCoiCalls_poly$sample)
allReports_sp = allReports %>%
select(sample, name, perBaseCoverageNormRounded) %>%
spread(name, perBaseCoverageNormRounded)
allReports_sp_mat = as.matrix(allReports_sp[,2:ncol(allReports_sp)])
rownames(allReports_sp_mat) = allReports_sp$sample
allReports_sp_mat[allReports_sp_mat >2] = 2
annotationTextSize = 20
topAnno = allReports %>%
filter(sample == .$sample[1]) %>%
select(name, extraField0) %>%
rename(`Gene Description` = extraField0) %>%
mutate(`Gene Description` = gsub(".*description=", "", `Gene Description`))%>%
mutate(`Gene Description` = gsub("\\].*", "", `Gene Description`)) %>%
mutate(`Gene Description` = ifelse(grepl("PHIST", `Gene Description`), gsub("\\).*", "", gsub(".*PHIST", "PHIST", `Gene Description`)), `Gene Description`))
topAnnoDf = topAnno %>%
select(-name) %>%
as.data.frame()
topAnnoColors = createColorListFromDf(topAnnoDf)
allReports_sp_mat_sampleOrder = hclust(dist(allReports_sp_mat))$order
allReports_sp_mat_sampleOrderDf = tibble(
sample = rownames(allReports_sp_mat)[allReports_sp_mat_sampleOrder],
order = 1:nrow(allReports_sp_mat)
#order = allReports_sp_mat_sampleOrder
)
metaReSelected = allMetaDeletionCalls[match(rownames(allReports_sp_mat), allMetaDeletionCalls$sample), ] %>%
left_join(allReports_sp_mat_sampleOrderDf)
rowAnno = tibble(metaReSelected[,c("sample","country","secondaryRegion", "order")]) %>%
rename(continent = secondaryRegion) %>%
mutate(`TARE1 Present On Chr13` = sample %in% hrp3Pat2_samplesWithTare1$sample) %>%
mutate(`Transposition With 5` = sample %in% samplesWithTransitionToChr5$sample) %>%
#select(-order)
#arrange(`TARE1 Present On Chr13`, `Transposition With 5`, order) %>%
arrange(`TARE1 Present On Chr13`, `Transposition With 5`, order) %>%
select(-order)
metaReSelected = metaReSelected[match(rowAnno$sample, metaReSelected$sample),]
allReports_sp_mat = allReports_sp_mat[match(rowAnno$sample, rownames(allReports_sp_mat)), ]
rowAnnoDf = rowAnno %>%
select(-sample) %>%
as.data.frame()
rowAnnoColors = createColorListFromDf(rowAnnoDf)
rowAnnoColors[["continent"]] = c("AFRICA" = "#E69F00",
"ASIA" = "#F0E442",
"OCEANIA" = "#F748A5",
"S_AMERICA" = "#0072B2")
create_dt(rowAnno %>% group_by(`TARE1 Present On Chr13`,`Transposition With 5`) %>% count() %>%
ungroup() %>%
mutate(total = sum(n)))Code
sideAnno = rowAnnotation(
df = rowAnnoDf,
col = rowAnnoColors,
gp = gpar(col = "grey10"),
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
gap = unit(0.1, "cm")
)
topAnno = HeatmapAnnotation(
df = topAnnoDf,
col = topAnnoColors,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
na_col = "#FFFFFF00"
)
library(circlize)
col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3)))
allReports_sp_mat_nolab = allReports_sp_mat
colnames(allReports_sp_mat_nolab) = NULL
allReports_sp_mat_hm = Heatmap(
allReports_sp_mat_nolab,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno,
right_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
cell_fun = function(j, i, x, y, width, height, fill) {
grid.rect(x = x, y = y, width = width, height = height *.9,
gp = gpar(fill = fill, col = NA))
},
rect_gp = gpar(type = "none"),
column_title = "Chr13[2,817kb:2,844kb]",
column_title_gp = gpar(fontsize = 20, fontface = "italic")
)
allReports_sp_mat_hm_noCluster = Heatmap(
allReports_sp_mat_nolab,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno,
right_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
cell_fun = function(j, i, x, y, width, height, fill) {
grid.rect(x = x, y = y, width = width, height = height *.9,
gp = gpar(fill = fill, col = NA))
},
rect_gp = gpar(type = "none"),
column_title = "Chr13[2,817kb:2,844kb]",
column_title_gp = gpar(fontsize = 20, fontface = "italic")
)Code
draw(allReports_sp_mat_hm, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", padding = unit(c(40, 2, 2, 2), "mm")) # , legend_border = T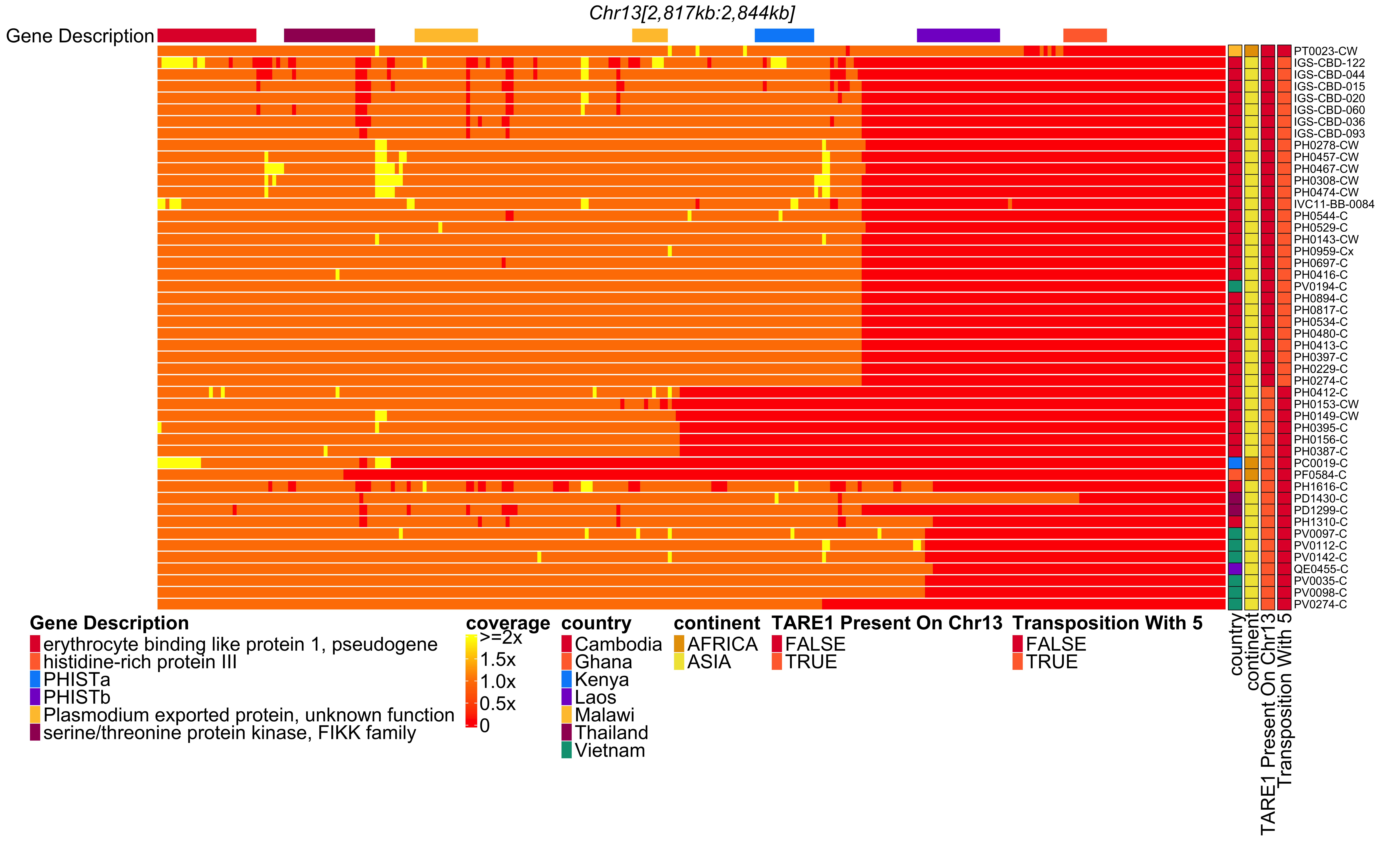
Code
pdf("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step100_windowSize100.pdf", useDingbats = F, width = 20, height = 12.5)
draw(allReports_sp_mat_hm, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", padding = unit(c(40, 2, 2, 2), "mm")) # , legend_border = T
dev.off()quartz_off_screen
2 Code
cairo_pdf("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step100_windowSize100_cairo.pdf", width = 20, height = 12.5)
draw(allReports_sp_mat_hm, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", padding = unit(c(40, 2, 2, 2), "mm")) # , legend_border = T
dev.off()quartz_off_screen
2