Code
cd /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq
nohup elucidator bamToFastq --bam ../bams/PfSD01.sorted.bam --out PfSD01 & Mapping the illumina data from PfHB3 and PfSD01 against their own assemblies to get estimations of coverage
cd /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq
nohup elucidator bamToFastq --bam ../bams/PfSD01.sorted.bam --out PfSD01 & unicycler -1 PfSD01_R1.fastq.gz -2 PfSD01_R2.fastq.gz -t 24 -o assemblies/unicycler_PfSD01 --verbosity 0 --no_pilon
unicycler -1 PfSD01_R1.fastq.gz -2 PfSD01_R2.fastq.gz -l /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/rawFastq/PfSD01.fastq.gz -t 24 -o assemblies/unicycler_PfSD01_withLong --verbosity 0 --no_pilon bwa mem -M -t 12 /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta PfSD01_R1.fastq.gz PfSD01_R2.fastq.gz| samtools sort -@ 12 -o alns/PfSD01-to-PfSD01.sorted.bam
bwa mem -M -t 12 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta PfHB3_R1.fastq.gz PfHB3_R2.fastq.gz| samtools sort -@ 12 -o alns/PfHB3-to-PfHB3.sorted.bam
bwa mem -M -t 12 /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/assemblies/PfHB3nano.fasta PfHB3_R1.fastq.gz PfHB3_R2.fastq.gz| samtools sort -@ 12 -o alns/PfHB3-to-PfHB3nano.sorted.bam elucidator fastaToBed --fasta /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta | elucidator bed3ToBed6 --bed STDIN | egrep PfSD01_11 | elucidator createWindowsInRegions --windowSize 50000 --step 25000 --bed STDIN > PfSD01_11_windows_size50000_step25000.bed
elucidator fastaToBed --fasta /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta | elucidator bed3ToBed6 --bed STDIN | egrep PfSD01_11 | elucidator createWindowsInRegions --windowSize 5000 --step 2500 --bed STDIN > PfSD01_11_windows_size5000_step2500.bed
elucidator fastaToBed --fasta /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta | elucidator bed3ToBed6 --bed STDIN | egrep PfSD01_11 | elucidator createWindowsInRegions --windowSize 1000 --step 500 --bed STDIN > PfSD01_11_windows_size1000_step500.bed
cd alns
echo PfSD01-to-PfSD01.sorted.bam > PfSD01_coverage_bams.txt
nohup elucidator bamMulticovBases --bams PfSD01_coverage_bams.txt --numThreads 10 --bedFnp ../PfSD01_11_windows_size50000_step25000.bed --out PfSD01_11_windows_size50000_step25000.tab.txt --overWrite &
nohup elucidator bamMulticovBases --bams PfSD01_coverage_bams.txt --numThreads 10 --bedFnp ../PfSD01_11_windows_size5000_step2500.bed --out PfSD01_11_windows_size5000_step2500.tab.txt --overWrite &elucidator fastaToBed --fasta /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta | elucidator bed3ToBed6 --bed STDIN | egrep PfSD01_13 | elucidator createWindowsInRegions --windowSize 50000 --step 25000 --bed STDIN > PfSD01_13_windows_size50000_step25000.bed
elucidator fastaToBed --fasta /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta | elucidator bed3ToBed6 --bed STDIN | egrep PfSD01_13 | elucidator createWindowsInRegions --windowSize 5000 --step 2500 --bed STDIN > PfSD01_13_windows_size5000_step2500.bed
elucidator fastaToBed --fasta /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta | elucidator bed3ToBed6 --bed STDIN | egrep PfSD01_13 | elucidator createWindowsInRegions --windowSize 1000 --step 500 --bed STDIN > PfSD01_13_windows_size1000_step500.bed
cd alns
echo PfSD01-to-PfSD01.sorted.bam > PfSD01_coverage_bams.txt
nohup elucidator bamMulticovBases --bams PfSD01_coverage_bams.txt --numThreads 10 --bedFnp ../PfSD01_13_windows_size50000_step25000.bed --out PfSD01_13_windows_size50000_step25000.tab.txt --overWrite &
nohup elucidator bamMulticovBases --bams PfSD01_coverage_bams.txt --numThreads 10 --bedFnp ../PfSD01_13_windows_size5000_step2500.bed --out PfSD01_13_windows_size5000_step2500.tab.txt --overWrite &nohup elucidatorlab BamGetBaseCovSpanningCov --bed ../PfSD01_11_windows_size5000_step2500.bed --sampName PfSD01 --genomeFnp /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta --numThreads 20 --bam PfSD01-to-PfSD01.sorted.bam --overWrite --out PfSD01_11_windows_size5000_step2500.tab.txt&
nohup elucidatorlab BamGetBaseCovSpanningCov --bed ../PfSD01_13_windows_size5000_step2500.bed --sampName PfSD01 --genomeFnp /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta --numThreads 20 --bam PfSD01-to-PfSD01.sorted.bam --overWrite --out PfSD01_13_windows_size5000_step2500.tab.txt &
nohup elucidatorlab BamGetBaseCovSpanningCov --bed ../PfSD01_11_windows_size1000_step500.bed --sampName PfSD01 --genomeFnp /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta --numThreads 20 --bam PfSD01-to-PfSD01.sorted.bam --overWrite --out PfSD01_11_windows_size1000_step500.tab.txt &
nohup elucidatorlab BamGetBaseCovSpanningCov --bed ../PfSD01_13_windows_size1000_step500.bed --sampName PfSD01 --genomeFnp /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta --numThreads 20 --bam PfSD01-to-PfSD01.sorted.bam --overWrite --out PfSD01_13_windows_size1000_step500.tab.txt &elucidator fastaToBed --fasta /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta | elucidator bed3ToBed6 --bed STDIN | egrep PfHB3_13 | elucidator createWindowsInRegions --windowSize 1000 --step 500 --bed STDIN > PfHB3_13_windows_size1000_step500.bed
elucidator fastaToBed --fasta /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta | elucidator bed3ToBed6 --bed STDIN | egrep PfHB3_11 | elucidator createWindowsInRegions --windowSize 1000 --step 500 --bed STDIN > PfHB3_11_windows_size1000_step500.bed
# PfHB3_00_0
elucidator fastaToBed --fasta /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta | elucidator bed3ToBed6 --bed STDIN | egrep PfHB3_00_0 | elucidator createWindowsInRegions --windowSize 1000 --step 500 --bed STDIN > PfHB3_00_0_windows_size1000_step500.bed
nohup elucidatorlab BamGetBaseCovSpanningCov --bed ../PfHB3_11_windows_size1000_step500.bed --sampName PfHB3 --genomeFnp /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta --numThreads 20 --bam PfHB3-to-PfHB3.sorted.bam --overWrite --out PfHB3_11_windows_size1000_step500.tab.txt &
nohup elucidatorlab BamGetBaseCovSpanningCov --bed ../PfHB3_13_windows_size1000_step500.bed --sampName PfHB3 --genomeFnp /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta --numThreads 20 --bam PfHB3-to-PfHB3.sorted.bam --overWrite --out PfHB3_13_windows_size1000_step500.tab.txt &
nohup elucidatorlab BamGetBaseCovSpanningCov --bed ../PfHB3_00_0_windows_size1000_step500.bed --sampName PfHB3 --genomeFnp /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta --numThreads 20 --bam PfHB3-to-PfHB3.sorted.bam --overWrite --out PfHB3_00_0_windows_size1000_step500.tab.txt &elucidator fastaToBed --fasta //tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/assemblies/PfHB3nano.fasta | elucidator bed3ToBed6 --bed STDIN | egrep PfHB3nano_13 | elucidator createWindowsInRegions --windowSize 1000 --step 500 --bed STDIN > PfHB3nano_13_windows_size1000_step500.bed
elucidator fastaToBed --fasta //tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/assemblies/PfHB3nano.fasta | elucidator bed3ToBed6 --bed STDIN | egrep PfHB3nano_11 | elucidator createWindowsInRegions --windowSize 1000 --step 500 --bed STDIN > PfHB3nano_11_windows_size1000_step500.bed
nohup elucidatorlab BamGetBaseCovSpanningCov --bed ../PfHB3nano_11_windows_size1000_step500.bed --sampName PfHB3 --genomeFnp //tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/assemblies/PfHB3nano.fasta --numThreads 20 --bam PfHB3-to-PfHB3nano.sorted.bam --overWrite --out PfHB3nano_11_windows_size1000_step500.tab.txt &
nohup elucidatorlab BamGetBaseCovSpanningCov --bed ../PfHB3nano_13_windows_size1000_step500.bed --sampName PfHB3 --genomeFnp //tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/assemblies/PfHB3nano.fasta --numThreads 20 --bam PfHB3-to-PfHB3nano.sorted.bam --overWrite --out PfHB3nano_13_windows_size1000_step500.tab.txt &all11 = tibble()
for(strain in c("PfSD01", "PfHB3")){
strain11 = readr::read_tsv(paste0("../../../mappingOutSurroundingRegions/endBeds/split_", strain, "_chrom11_toEnd_genes.tab.txt")) %>%
mutate(strain = strain ) %>%
mutate(chromGlobal = gsub(paste0(strain, "_"), "", col.0)) %>%
mutate(chromGlobal = gsub("_v3", "", chromGlobal)) %>%
rename(chrom = col.0,
start = col.1,
end = col.2)
all11 = bind_rows(all11, strain11)
}
all11 = all11 %>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse("membrane associated erythrocyte binding-like protein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>%
mutate(description = ifelse("membrane associated erythrocyte binding-likeprotein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>%
mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description)) %>%
mutate(description = gsub(",putative", ", putative", description)) %>%
mutate(description = gsub(",pseudogene", ", pseudogene", description))%>%
mutate(description = gsub("unknownfunction", "unknown function", description))%>%
mutate(description = gsub("conserved protein, unknown function", "conserved Plasmodium protein, unknown function", description)) %>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description))%>%
mutate(description = gsub(",pseudogene", ", pseudogene", description))%>%
mutate(description = gsub("unknownfunction", "unknown function", description))%>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse(grepl("sporozoite and liver stage tryptophan-rich protein, putative", description), "tryptophan/threonine-rich antigen", description))%>%
mutate(description = ifelse(grepl("CRA domain-containing protein, putative", description), "conserved Plasmodium protein, unknown function", description))%>%
mutate(description = gsub(",pseudogene", ", pseudogene", description)) %>%
mutate(description = gsub("surfaceantigen", "surface antigen", description)) %>%
mutate(description = gsub("Tetratricopeptide repeat, putative", "tetratricopeptide repeat protein, putative", description)) %>%
mutate(description = gsub("transmembraneprotein", "transmembrane protein", description)) %>%
mutate(description = ifelse(grepl("PfEMP1", description) & grepl("pseudogene", description), "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>%
mutate(description = gsub("PIR protein", "stevor", description)) %>%
mutate(description = gsub("erythrocyte membrane protein 1-like", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>%
mutate(description = gsub("acidic terminal segments, variant surface antigen of PfEMP1, putative", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description))%>%
mutate(description = ifelse(grepl("CoA binding protein", description, ignore.case = T), "acyl-CoA binding protein", description)) %>%
mutate(description = ifelse(grepl("transfer RNA", description) | grepl("tRNA", description), "tRNA", description))%>%
mutate(description = ifelse(grepl("cytoadherence", description), "CLAG", description))%>%
mutate(description = ifelse(grepl("surface-associated interspersed protein", description), "SURFIN", description))%>%
mutate(description = ifelse(grepl("SURFIN", description), "SURFIN", description))%>%
mutate(description = ifelse(grepl("stevor-like", description), "stevor, pseudogene", description)) %>%
mutate(description = ifelse(grepl("exported protein family", description), "exported protein family", description)) %>%
mutate(description = ifelse(grepl("ribosomal RNA", description), "rRNA", description)) %>%
mutate(description = ifelse(grepl("serine/threonine protein kinase", description), "serine/threonine protein kinase, FIKK family", description)) %>%
mutate(description = ifelse(grepl("hypothetical protein", description), "hypothetical protein, conserved", description)) %>%
mutate(description = ifelse(grepl("conserved Plasmodium protein, unknown function", description), "hypothetical protein, conserved", description)) %>%
mutate(description = ifelse(grepl("Rifin/stevor family, putative", description), "stevor", description)) %>%
mutate(description = ifelse(grepl("stevor", description), "stevor", description)) %>%
mutate(description = ifelse(grepl("rifin", description), "rifin", description)) %>%
mutate(description = ifelse(grepl("erythrocyte membrane protein 1 (PfEMP1), pseudogene", description), "erythrocyte membrane protein 1 (PfEMP1)", description)) %>%
mutate(description = ifelse(grepl("erythrocyte membrane protein 1", description), "erythrocyte membrane protein 1 (PfEMP1)", description)) %>%
mutate(description = ifelse(grepl("probably protein", description), "unspecified product", description)) %>%
mutate(description = ifelse(grepl("RESA", description), "RESA", description)) %>%
mutate(description = ifelse(grepl("ring-infected erythrocyte surface antigen", description), "ring-infected erythrocyte surface antigen", description))%>%
mutate(description = ifelse(grepl("Duffy binding domain/Erythrocyte binding antigen175, putative", description), "erythrocyte binding like protein 1", description))%>%
mutate(description = ifelse(grepl("erythrocyte binding like protein 1", description), "erythrocyte binding like protein 1", description)) %>%
mutate(description = ifelse(grepl("unspecified product", description), "hypothetical protein, conserved", description))%>%
mutate(description = ifelse(grepl("probable protein, unknown function", description), "hypothetical protein, conserved", description))%>%
mutate(description = ifelse(grepl("RESA", description), "ring-infected erythrocyte surface antigen", description)) all13 = tibble()
for(strain in c("PfSD01", "PfHB3")){
strain13 = readr::read_tsv(paste0("../../../mappingOutSurroundingRegions/endBeds/split_", strain, "_chrom13_toEnd_genes.tab.txt")) %>%
mutate(strain = strain ) %>%
mutate(chromGlobal = gsub(paste0(strain, "_"), "", col.0)) %>%
mutate(chromGlobal = gsub("_v3", "", chromGlobal)) %>%
rename(chrom = col.0,
start = col.1,
end = col.2)
all13 = bind_rows(all13, strain13)
}
all13 = all13 %>%
filter(col.3 %!in% c("Pf3D7_13_v3_2794236_2794851_PF3D7_1370500", "Pf3D7_13_v3_2796118_2797144_PF3D7_1370800", "Pf3D7_13_v3_2797506_2798103_PF3D7_1370900")) %>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse("membrane associated erythrocyte binding-like protein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>%
mutate(description = ifelse("membrane associated erythrocyte binding-likeprotein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>%
mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description)) %>%
mutate(description = gsub(",putative", ", putative", description)) %>%
mutate(description = gsub(",pseudogene", ", pseudogene", description))%>%
mutate(description = gsub("unknownfunction", "unknown function", description))%>%
mutate(description = gsub("conserved protein, unknown function", "conserved Plasmodium protein, unknown function", description)) %>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description))%>%
mutate(description = gsub(",pseudogene", ", pseudogene", description))%>%
mutate(description = gsub("unknownfunction", "unknown function", description))%>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse(grepl("sporozoite and liver stage tryptophan-rich protein, putative", description), "tryptophan/threonine-rich antigen", description))%>%
mutate(description = ifelse(grepl("CRA domain-containing protein, putative", description), "conserved Plasmodium protein, unknown function", description))%>%
mutate(description = gsub(",pseudogene", ", pseudogene", description)) %>%
mutate(description = gsub("surfaceantigen", "surface antigen", description)) %>%
mutate(description = gsub("Tetratricopeptide repeat, putative", "tetratricopeptide repeat protein, putative", description)) %>%
mutate(description = gsub("transmembraneprotein", "transmembrane protein", description)) %>%
mutate(description = ifelse(grepl("PfEMP1", description) & grepl("pseudogene", description), "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>%
mutate(description = gsub("PIR protein", "stevor", description)) %>%
mutate(description = gsub("erythrocyte membrane protein 1-like", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>%
mutate(description = gsub("acidic terminal segments, variant surface antigen of PfEMP1, putative", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description))%>%
mutate(description = ifelse(grepl("CoA binding protein", description, ignore.case = T), "acyl-CoA binding protein", description)) %>%
mutate(description = ifelse(grepl("transfer RNA", description) | grepl("tRNA", description), "tRNA", description))%>%
mutate(description = ifelse(grepl("cytoadherence", description), "CLAG", description))%>%
mutate(description = ifelse(grepl("surface-associated interspersed protein", description), "SURFIN", description))%>%
mutate(description = ifelse(grepl("SURFIN", description), "SURFIN", description))%>%
mutate(description = ifelse(grepl("stevor-like", description), "stevor, pseudogene", description)) %>%
mutate(description = ifelse(grepl("exported protein family", description), "exported protein family", description)) %>%
mutate(description = ifelse(grepl("ribosomal RNA", description), "rRNA", description)) %>%
mutate(description = ifelse(grepl("serine/threonine protein kinase", description), "serine/threonine protein kinase, FIKK family", description)) %>%
mutate(description = ifelse(grepl("hypothetical protein", description), "hypothetical protein, conserved", description)) %>%
mutate(description = ifelse(grepl("conserved Plasmodium protein, unknown function", description), "hypothetical protein, conserved", description)) %>%
mutate(description = ifelse(grepl("Rifin/stevor family, putative", description), "stevor", description)) %>%
mutate(description = ifelse(grepl("stevor", description), "stevor", description)) %>%
mutate(description = ifelse(grepl("rifin", description), "rifin", description)) %>%
mutate(description = ifelse(grepl("erythrocyte membrane protein 1 (PfEMP1), pseudogene", description), "erythrocyte membrane protein 1 (PfEMP1)", description)) %>%
mutate(description = ifelse(grepl("erythrocyte membrane protein 1", description), "erythrocyte membrane protein 1 (PfEMP1)", description)) %>%
mutate(description = ifelse(grepl("probably protein", description), "unspecified product", description)) %>%
mutate(description = ifelse(grepl("RESA", description), "RESA", description)) %>%
mutate(description = ifelse(grepl("ring-infected erythrocyte surface antigen", description), "ring-infected erythrocyte surface antigen", description))%>%
mutate(description = ifelse(grepl("Duffy binding domain/Erythrocyte binding antigen175, putative", description), "erythrocyte binding like protein 1", description))%>%
mutate(description = ifelse(grepl("erythrocyte binding like protein 1", description), "erythrocyte binding like protein 1", description)) %>%
mutate(description = ifelse(grepl("unspecified product", description), "hypothetical protein, conserved", description))%>%
mutate(description = ifelse(grepl("probable protein, unknown function", description), "hypothetical protein, conserved", description))%>%
mutate(description = ifelse(grepl("RESA", description), "ring-infected erythrocyte surface antigen", description))
allRepeats_filt_starts_ends_tares = readr::read_tsv("../surroundingRegionsMaterials/alltrfs/allRepeats_filt_starts_ends_tares.tsv") %>%
mutate(start = X2,
end = X3,
chrom = X1)descriptionColorsNames = unique(c(all13$description, all11$description))
rawDescriptionColors = scheme$hex(length(descriptionColorsNames))
names(rawDescriptionColors) = descriptionColorsNames
genomicElementsColors = c()
genomicElementsColors["TARE1"] = "#5A81AB"
genomicElementsColors["SB3"] = "#B483A7"
rawGeneAnnotationsColors = rawDescriptionColors
descriptionColors = c(rawDescriptionColors, genomicElementsColors)
geneAnnotationsColors = descriptionColorsPlotting across the whole of chromosome 11 in 1kb windows stepping very 500 bases
PfSD01_11_windows_size1000_step500 = readr::read_tsv("data/PfSD01_11_windows_size1000_step500.tab.txt")%>%
mutate(medianPerBaseCov = median(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = perBaseCoverage/medianPerBaseCov) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNorm / 0.5)*0.5)
ggplot(PfSD01_11_windows_size1000_step500) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage,
fill = factor(perBaseCoverageNormRounded)
)) + scale_fill_tableau(name = "Normalized Coverage") + sofonias_theme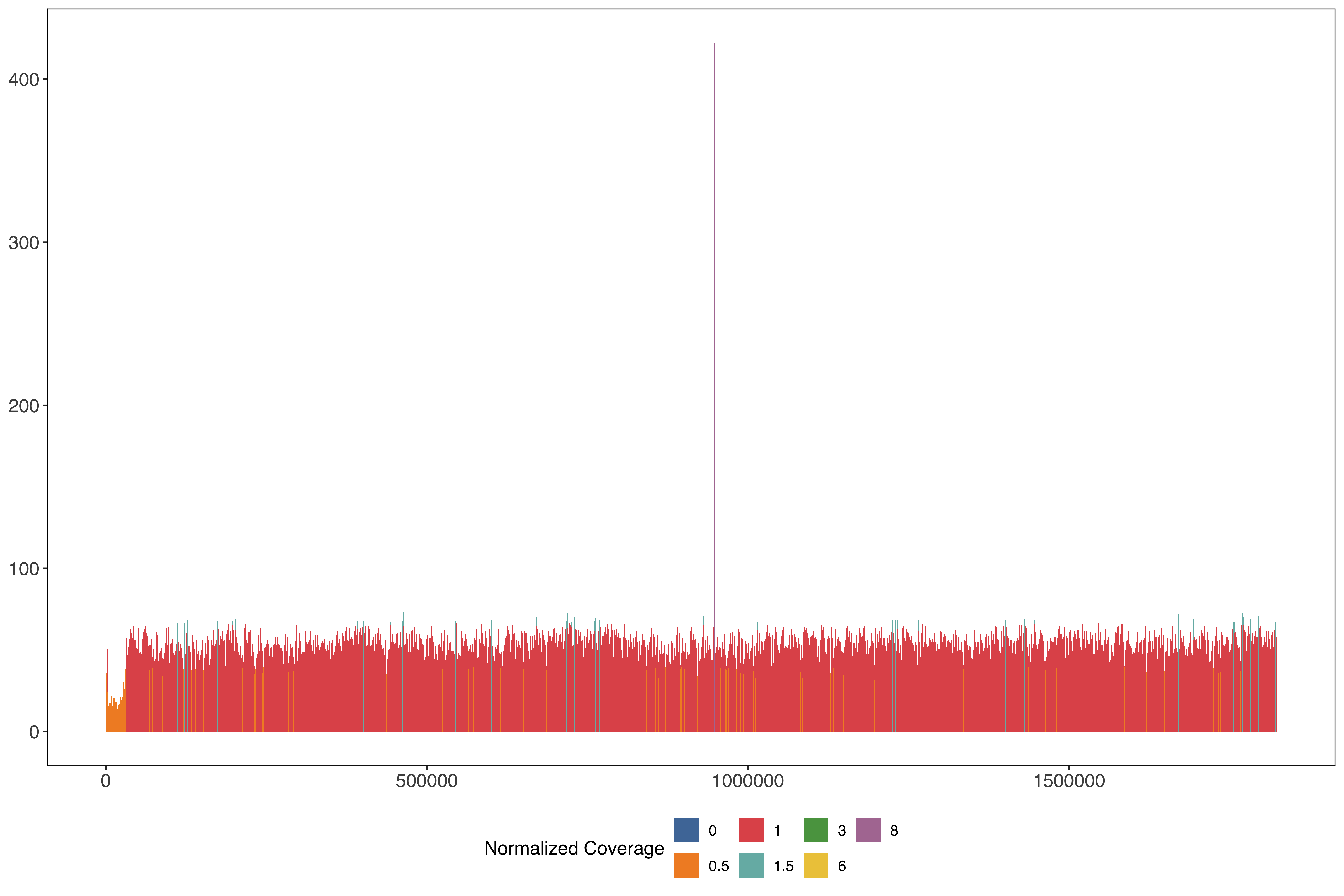
PfSD01_11_windows_size1000_step500_filt = PfSD01_11_windows_size1000_step500 %>%
filter(start >= min(all11$start))
allRepeats_filt_starts_ends_tares_chrom11 = allRepeats_filt_starts_ends_tares %>%
filter(grepl("_11", X1)) %>%
filter(X2 >= min(all11$start))
all11_sd01 = all11 %>% filter(strain == "PfSD01")
PfSD01_chrom11_plot = ggplot(PfSD01_11_windows_size1000_step500_filt) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage
)) + geom_rect(
data = all11_sd01,
aes(
xmin = start,
xmax = end,
ymin = -25,
ymax = 0,
fill = description
),
color = "black"
)
if(nrow(allRepeats_filt_starts_ends_tares_chrom11 %>% filter(strain == "PfSD01")) > 0){
PfSD01_chrom11_plot = PfSD01_chrom11_plot+
geom_rect(
data = allRepeats_filt_starts_ends_tares_chrom11 %>% filter(strain == "PfSD01"),
aes(
xmin = start,
xmax = end,
ymin = -25/2,
ymax = 0,
fill = repeatType,
color = repeatType
)
)
}
PfSD01_chrom11_plot = PfSD01_chrom11_plot+
sofonias_theme +
scale_fill_manual(values = descriptionColors[names(descriptionColors) %in% c(all11$description, "SB3", "TARE1")]) +
labs(title = "Illumina Coverage PfSD01 Pacbio assembly Chr11")
ggplotly(PfSD01_chrom11_plot)PfSD01_chrom11_plot = PfSD01_chrom11_plot +
scale_fill_manual(
"Genes",
values = geneAnnotationsColors,
breaks = names(rawGeneAnnotationsColors),
labels = names(rawGeneAnnotationsColors),
guide = guide_legend(
override.aes = list(
color = rawGeneAnnotationsColors[unique(all11_sd01$description)],
fill = rawGeneAnnotationsColors[unique(all11_sd01$description)],
alpha = 1,
shape = 22,
size = 5
),
nrow = 4,
order = 1
)
) +
scale_color_manual(
"Genomic\nElements",
# values = geneAnnotationsColors,
values = c("black", "black"),
breaks = names(genomicElementsColors),
labels = names(genomicElementsColors),
guide = guide_legend(
override.aes = list(
color = genomicElementsColors,
fill = genomicElementsColors,
alpha = 1,
shape = 22,
size = 5
),
ncol = 1,
order = 2
)
)
pdf("PfSD01_illumina_against_pacbio_chrom11_plot.pdf", width = 17.5, height = 8, useDingbats = F)
print(PfSD01_chrom11_plot)
dev.off()quartz_off_screen
2 Plotting across the whole of chromosome 13 in 1kb windows stepping very 500 bases
PfSD01_13_windows_size1000_step500 = readr::read_tsv("data/PfSD01_13_windows_size1000_step500.tab.txt")%>%
mutate(medianPerBaseCov = median(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = perBaseCoverage/medianPerBaseCov) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNorm / 0.5)*0.5)
ggplot(PfSD01_13_windows_size1000_step500) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage,
fill = factor(perBaseCoverageNormRounded)
)) + scale_fill_tableau(name = "Normalized Coverage") + sofonias_theme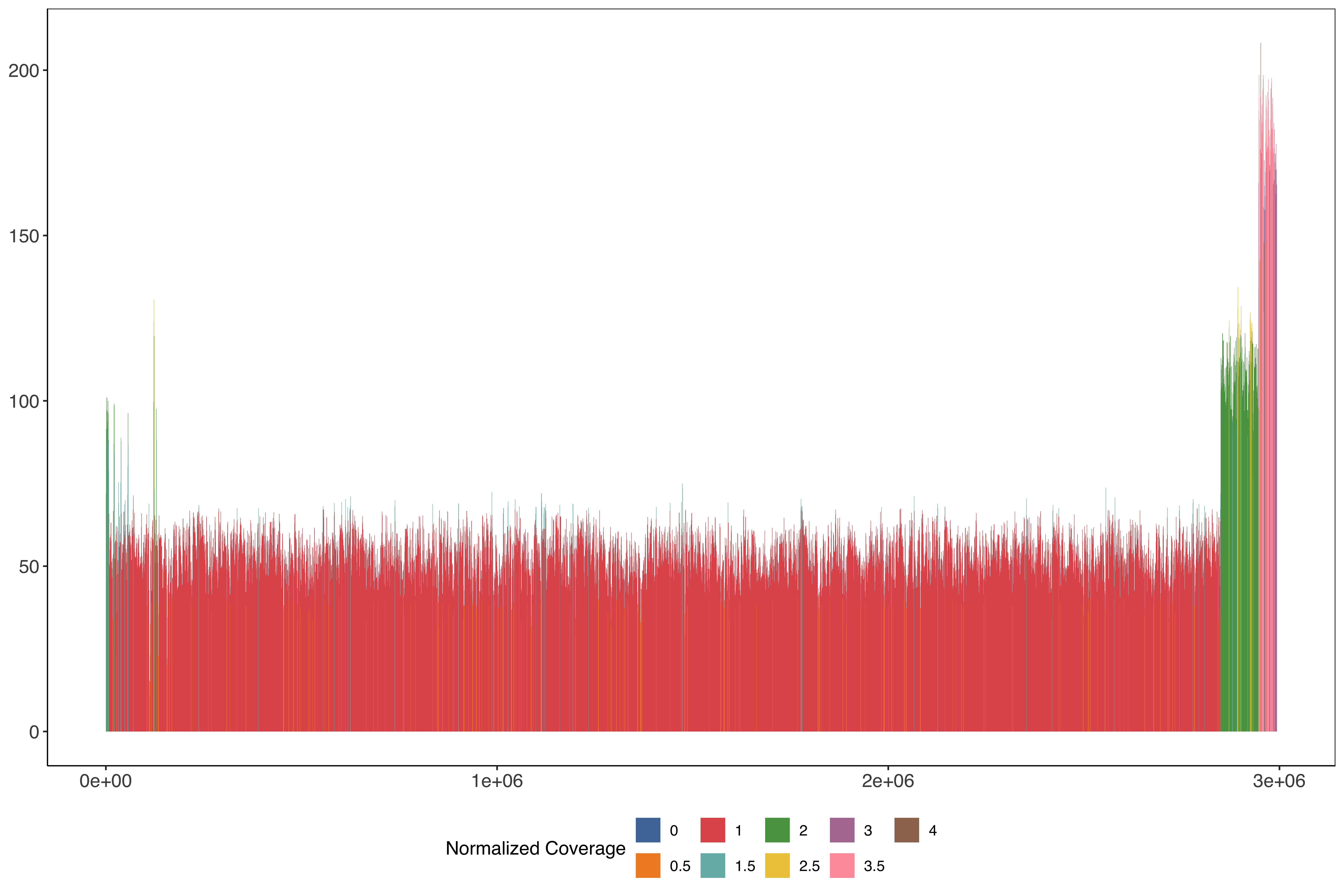
cd ../surroundingRegionsMaterials/alltrfs/trf_PfSD01/
# create a bed file fromt the trf output
# sort and merge
elucidator bedCoordSort --bed combined.bed | bedtools merge | elucidator bed3ToBed6 --bed STDIN --out merged_sorted_combined.bed --overWrite
# filter by length
elucidator filterBedRecordsByLength --bed merged_sorted_combined.bed --minLen 50 --out filtered_lenGreater50_merged_sorted_combined.bed --overWriteSub-select repeats based off of size based on their likelihood to slip during amplification
isPossibleDiRepeat <-function(repeatUnit){
if(2 == nchar(repeatUnit)){
return (TRUE)
}else if(0 == nchar(repeatUnit)%%2 ){
isDi = T
front = substr(repeatUnit, 1,2)
#print(paste0(repeatUnit, ",", nchar(repeatUnit)))
for(pos in seq(3,nchar(repeatUnit),2)){
if(substr(repeatUnit, pos,pos + 1) != front){
isDi = F
break;
}
}
return(isDi)
}
return(FALSE)
}
isPossibleTriRepeat <-function(repeatUnit){
if(3 == nchar(repeatUnit)){
return (TRUE)
}else if(0 == nchar(repeatUnit)%%3 ){
isTri = T
front = substr(repeatUnit, 1,3)
#print(paste0(repeatUnit, ",", nchar(repeatUnit)))
for(pos in seq(4,nchar(repeatUnit),3)){
if(substr(repeatUnit, pos, pos + 2) != front){
isTri = F
break;
}
}
return(isTri)
}
return(FALSE)
}
combinedTandemRepeats = readr::read_tsv("../surroundingRegionsMaterials/alltrfs/trf_PfSD01/combined.bed", col_names = F) %>%
mutate(repeatCode = gsub(".*__", "", X4)) %>%
mutate(repeatUnit = gsub("_.*", "", repeatCode)) %>%
mutate(repeatUnitSize = nchar(repeatUnit)) %>%
mutate(repeatNumber = as.numeric(gsub(".*_x", "", repeatCode))) %>%
group_by(X1,X2,X3,X4,X5,X6) %>%
mutate(isDi = isPossibleDiRepeat(repeatUnit))%>%
mutate(isTri = isPossibleTriRepeat(repeatUnit)) %>%
group_by()
combinedTandemRepeats = combinedTandemRepeats %>%
mutate(repeatToAvoid = (repeatUnitSize == 1 & X5 >10) | (isDi & X5 >= 12) | (isTri & X5 >=21) | X5 >=50)combinedTandemRepeats_toAvoid = combinedTandemRepeats %>%
filter(repeatToAvoid)
write_tsv(combinedTandemRepeats_toAvoid %>%
select(1:6), "../surroundingRegionsMaterials/alltrfs/trf_PfSD01/repeatsToAvoid.bed", col_names = F)cd ../surroundingRegionsMaterials/alltrfs/trf_PfSD01/
cat repeatsToAvoid.bed filtered_lenGreater50_merged_sorted_combined.bed | elucidator bedCoordSort --bed STDIN | bedtools merge | elucidator bed3ToBed6 --bed STDIN | elucidator bedCoordSort --bed STDIN --out finalTandemsToAvoid.bed --overWrite
elucidator getInterveningRegions --bed finalTandemsToAvoid.bed --genome /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta --addMissingChromosomes --padding 20 | elucidator bed3ToBed6 --bed STDIN | elucidator filterBedRecordsByLength --bed STDIN --minLen 100 --out inbetweenLargeTandems.bed --overWrite
elucidator getOverlappingBedRegions --bed ../alltrfs/trf_PfSD01/inbetweenLargeTandems.bed --intersectWithBed PfSD01_chroms_13_ending.bed | cut -f1-6 | elucidator bedGetIntersectingGenesInGff --gff /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/info/gff/PfSD01.gff --bed STDIN --overWrite --out inbetweenLargeTandems_PfSD01_chroms_13_ending.bedPfSD01_chroms = readr::read_tsv("../../../mappingOutSurroundingRegions/chromLengths/PfSD01.txt", col_names = c("chrom", "length"))
PfSD01_chroms_13 = PfSD01_chroms%>%
filter(chrom == "PfSD01_13")
PfSD01_chroms_13_ending = PfSD01_chroms_13 %>%
mutate(start= min(all13$start)) %>%
rename(end = length) %>%
select(chrom, start, end) %>%
mutate(name = paste0(chrom, "-", start, "-", end)) %>%
mutate(len = end - start,
strand = "+") %>%
rename(`#chrom` = chrom)
write_tsv(PfSD01_chroms_13_ending, "PfSD01_chroms_13_ending.bed")
PfSD01_chroms_11 = PfSD01_chroms%>%
filter(chrom == "PfSD01_11")
PfSD01_chroms_11_ending = PfSD01_chroms_11 %>%
mutate(start= min(all11$start)) %>%
rename(end = length) %>%
select(chrom, start, end) %>%
mutate(name = paste0(chrom, "-", start, "-", end)) %>%
mutate(len = end - start,
strand = "+") %>%
rename(`#chrom` = chrom)
write_tsv(PfSD01_chroms_11_ending, "PfSD01_chroms_11_ending.bed")PfSD01_13_windows_size1000_step500_filt = PfSD01_13_windows_size1000_step500 %>%
filter(start >= min(all13$start))
allRepeats_filt_starts_ends_tares_chrom13 = allRepeats_filt_starts_ends_tares %>%
filter(grepl("_13", X1)) %>%
filter(X2 >= min(all13$start))
all13_sd01 = all13 %>% filter(strain == "PfSD01")
PfSD01_chrom13_plot = ggplot(PfSD01_13_windows_size1000_step500_filt) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage
)) + geom_rect(
data = all13_sd01,
aes(
xmin = start,
xmax = end,
ymin = -25,
ymax = 0,
fill = description
),
color = "black"
)
if(nrow(allRepeats_filt_starts_ends_tares_chrom13 %>% filter(strain == "PfSD01")) > 0){
PfSD01_chrom13_plot = PfSD01_chrom13_plot+
geom_rect(
data = allRepeats_filt_starts_ends_tares_chrom13 %>% filter(strain == "PfSD01"),
aes(
xmin = start,
xmax = end,
ymin = -25/2,
ymax = 0,
fill = repeatType,
color = repeatType
)
)
}
PfSD01_chrom13_plot = PfSD01_chrom13_plot+
sofonias_theme +
scale_fill_manual(values = descriptionColors[names(descriptionColors) %in% c(all13$description, "SB3", "TARE1")])+
scale_color_manual(values = descriptionColors[names(descriptionColors) %in% c(all13$description, "SB3", "TARE1")])+
labs(title = "Illumina Coverage PfSD01 Pacbio assembly Chr13")
ggplotly(PfSD01_chrom13_plot)PfSD01_chrom13_plot = PfSD01_chrom13_plot +
scale_fill_manual(
"Genes",
values = geneAnnotationsColors,
breaks = names(rawGeneAnnotationsColors),
labels = names(rawGeneAnnotationsColors),
guide = guide_legend(
override.aes = list(
color = rawGeneAnnotationsColors[unique(all13_sd01$description)],
fill = rawGeneAnnotationsColors[unique(all13_sd01$description)],
alpha = 1,
shape = 22,
size = 5
),
nrow = 5,
order = 1
)
) +
scale_color_manual(
"Genomic\nElements",
# values = geneAnnotationsColors,
values = c("black", "black"),
breaks = names(genomicElementsColors),
labels = names(genomicElementsColors),
guide = guide_legend(
override.aes = list(
color = genomicElementsColors,
fill = genomicElementsColors,
alpha = 1,
shape = 22,
size = 5
),
ncol = 1,
order = 2
)
)
pdf("PfSD01_illumina_against_pacbio_chrom13_plot.pdf", width = 17.5, height = 8, useDingbats = F)
print(PfSD01_chrom13_plot)
dev.off()quartz_off_screen
2 In between tandems
cd /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq
PathWeaver BamExtractPathwaysFromRegion --genomeDir /tank/data/genomes/plasmodium/genomes/pf_others/genomes/ --primaryGenome PfSD01 --bed /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/PfSD01_beds/inbetweenLargeTandems_PfSD01_chroms_13_ending.bed --bam alns/PfSD01-to-PfSD01.sorted.bam --dout inbetweenLargeTandems_PfSD01_chroms_13_ending/PfSD01-to-PfSD01_inbetweenLargeTandems_PfSD01_chroms_13_ending --numThreads 40 --overWriteDir --writeOutFinalDot --keepTemporaryFiles --bamExtractTrimToRegionbasicInfo = readr::read_tsv("data/PfSD01-to-PfSD01_inbetweenLargeTandems_PfSD01_chroms_13_ending/final/basicInfoPerRegion.tab.txt") %>%
mutate(perBaseCoverageNorm = perBaseCoverage/mean(PfSD01_13_windows_size1000_step500$medianPerBaseCov)) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNorm / 0.5)*0.5)
PfSD01_chrom13_plot = ggplot(basicInfo) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage,
perBaseCoverage = perBaseCoverage,
fill = factor(perBaseCoverageNormRounded)
)) + geom_rect(
data = all13 %>% filter(strain == "PfSD01"),
aes(
xmin = start,
xmax = end,
ymin = -25,
ymax = 0,
fill = description
),
color = "black"
)
if(nrow(allRepeats_filt_starts_ends_tares_chrom13 %>% filter(strain == "PfSD01")) > 0){
PfSD01_chrom13_plot = PfSD01_chrom13_plot+
geom_rect(
data = allRepeats_filt_starts_ends_tares_chrom13 %>% filter(strain == "PfSD01"),
aes(
xmin = start,
xmax = end,
ymin = -25/2,
ymax = 0,
fill = repeatType,
color = repeatType
)
)
}
coverages = unique(basicInfo$perBaseCoverageNormRounded)
library(circlize)
col_fun = colorRamp2(c(0, median(coverages), max(coverages)), c(heat.colors(3)))
coveragesCols = col_fun(coverages)
names(coveragesCols) = coverages
PfSD01_chrom13_plot = PfSD01_chrom13_plot+
sofonias_theme +
scale_fill_manual(values = c(coveragesCols, descriptionColors[names(descriptionColors) %in% c(all13$description, "SB3", "TARE1")]) )+
scale_color_manual(values = c(coveragesCols, descriptionColors[names(descriptionColors) %in% c(all13$description, "SB3", "TARE1")]) )+
labs(title = "Illumina Coverage PfSD01 Pacbio assembly Chr13")
ggplotly(PfSD01_chrom13_plot)elucidator bamToBed --bam bamsMinimap2AgainstPfSD01/PfSD01PermethION.sorted.bam | elucidator getOverlappingBedRegions --bed STDIN --intersectWithBed ../PfSD01_beds/PfSD01_chroms_13_ending.bed > PfSD01PermethION_chrom13_ending.bed
elucidator bedAddSmartIDForPlotting --bed PfSD01PermethION_chrom13_ending.bed --overWrite --out PfSD01PermethION_chrom13_ending_withPlotID.bedchrom13_ending = readr::read_tsv("../spanningReads/PfSD01PermethION_chrom13_ending_withPlotID.bed", col_names = F) %>%
mutate(start = X2,
end = X3,
len = X5)
chrom13_ending_nanopore_plot = ggplot(chrom13_ending) +
geom_rect(aes(xmin = X2, xmax = X3,
ymin = X8, ymax = X8 + 1,
len = len,
start = start,
end = end)) +
sofonias_theme + geom_rect(
data = all13 %>% filter(strain == "PfSD01"),
aes(
xmin = start,
xmax = end,
ymin = -25,
ymax = 0,
fill = description
),
color = "black"
)
if(nrow(allRepeats_filt_starts_ends_tares_chrom13 %>% filter(strain == "PfSD01")) > 0){
chrom13_ending_nanopore_plot = chrom13_ending_nanopore_plot+
geom_rect(
data = allRepeats_filt_starts_ends_tares_chrom13 %>% filter(strain == "PfSD01"),
aes(
xmin = start,
xmax = end,
ymin = -25/2,
ymax = 0,
fill = repeatType,
color = repeatType
)
)
}
chrom13_ending_nanopore_plot = chrom13_ending_nanopore_plot+
sofonias_theme +
scale_fill_manual(values = descriptionColors[names(descriptionColors) %in% c(all13$description, "SB3", "TARE1")])+
scale_color_manual(values = descriptionColors[names(descriptionColors) %in% c(all13$description, "SB3", "TARE1")])+
labs(title = "Nanopore Coverage PfSD01 Pacbio assembly Chr13")ggplotly(chrom13_ending_nanopore_plot)minimap2 -x map-ont -t 30 -a /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta canuoutput_sd01_promethionMinION.contigs.fasta | samtools sort --threads 30 -o canuoutput_sd01_promethionMinION.contigs.fasta.sorted.bam && samtools index canuoutput_sd01_promethionMinION.contigs.fasta.sorted.bam
elucidator bamToBed --bam canuoutput_sd01_promethionMinION.contigs.fasta.sorted.bam | elucidator getOverlappingBedRegions --bed STDIN --intersectWithBed ../../PfSD01_beds/PfSD01_chroms_13_ending.bed > canuoutput_sd01_promethionMinION.contigs.fasta_chrom13_ending.bed
elucidator bedAddSmartIDForPlotting --bed canuoutput_sd01_promethionMinION.contigs.fasta_chrom13_ending.bed --overWrite --out canuoutput_sd01_promethionMinION.contigs.fasta_chrom13_ending_withPlotID.bed
elucidator bamToBed --bam canuoutput_sd01_promethionMinION.contigs.fasta.sorted.bam | elucidator getOverlappingBedRegions --bed STDIN --intersectWithBed ../../PfSD01_beds/PfSD01_chroms_11_ending.bed > canuoutput_sd01_promethionMinION.contigs.fasta_chrom11_ending.bed
elucidator bedAddSmartIDForPlotting --bed canuoutput_sd01_promethionMinION.contigs.fasta_chrom11_ending.bed --overWrite --out canuoutput_sd01_promethionMinION.contigs.fasta_chrom11_ending_withPlotID.bednucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta canuoutput_sd01_promethionMinION.contigs.fasta --prefix canuoutput_sd01_promethionMinION.contigs_to_PfSD01_nucmer
show-coords -T -l -c -H canuoutput_sd01_promethionMinION.contigs_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out canuoutput_sd01_promethionMinION.contigs_to_PfSD01_nucmer.delta.bed
elucidator splitColumnContainingMeta --file canuoutput_sd01_promethionMinION.contigs_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out canuoutput_sd01_promethionMinION.contigs_to_PfSD01_nucmer.delta.tsv
nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta canuoutput_sd01_promethionMinION.contigs.fasta --prefix canuoutput_sd01_promethionMinION.contigs_to_Pf3D7_nucmer
show-coords -T -l -c -H canuoutput_sd01_promethionMinION.contigs_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out canuoutput_sd01_promethionMinION.contigs_to_Pf3D7_nucmer.delta.bed
elucidator splitColumnContainingMeta --file canuoutput_sd01_promethionMinION.contigs_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out canuoutput_sd01_promethionMinION.contigs_to_Pf3D7_nucmer.delta.tsvchrom13_ending = readr::read_tsv("canuoutput_sd01_promethionMinION.contigs.fasta_chrom13_ending_withPlotID.bed", col_names = F) %>%
mutate(start = X2,
end = X3,
len = X5,
name = X4)
chrom13_ending_nanopore_plot = ggplot(chrom13_ending) +
geom_rect(aes(xmin = X2, xmax = X3,
ymin = X8, ymax = X8 + 1,
len = len,
start = start,
end = end,
name = name
)) +
sofonias_theme + geom_rect(
data = all13 %>% filter(strain == "PfSD01"),
aes(
xmin = start,
xmax = end,
ymin = -25,
ymax = 0,
fill = description
),
color = "black"
)
if(nrow(allRepeats_filt_starts_ends_tares_chrom13 %>% filter(strain == "PfSD01")) > 0){
chrom13_ending_nanopore_plot = chrom13_ending_nanopore_plot+
geom_rect(
data = allRepeats_filt_starts_ends_tares_chrom13 %>% filter(strain == "PfSD01"),
aes(
xmin = start,
xmax = end,
ymin = -25/2,
ymax = 0,
fill = repeatType,
color = repeatType
)
)
}
chrom13_ending_nanopore_plot = chrom13_ending_nanopore_plot+
sofonias_theme +
scale_fill_manual(values = descriptionColors[names(descriptionColors) %in% c(all13$description, "SB3", "TARE1")])+
scale_color_manual(values = descriptionColors[names(descriptionColors) %in% c(all13$description, "SB3", "TARE1")])+
labs(title = "Nanopore Assembly Coverage PfSD01 Pacbio assembly Chr13")ggplotly(chrom13_ending_nanopore_plot)nanopore_nucmer_results = readr::read_tsv("canuoutput_sd01_promethionMinION.contigs_to_PfSD01_nucmer.delta.tsv")%>%
mutate(id = row_number(),
start = col.1,
end = col.2,
name = col.3,
strand = col.5,
chrom = col.0,
mapLength = col.4,
actualLength = actualEnd - actualStart)
nanopore_nucmer_results_tig00000054 = nanopore_nucmer_results %>%
filter(col.4 > 1000) %>%
filter(col.3 == "tig00000054") %>%
arrange(desc(perID), desc(col.4) ) %>%
mutate(id = row_number())
ggplotly(ggplot(nanopore_nucmer_results_tig00000054) +
geom_rect(aes(xmin = actualStart, xmax = actualEnd,
ymin = id, ymax = id + 1,
fill = col.0,
chrom = chrom,
start = start,
end = end,
mapLength = mapLength,
name = name,
actualStart = actualStart,
actualEnd = actualEnd,
actualLength = actualLength,
strand = strand)) +
scale_fill_tableau("Tableau 20") +
sofonias_theme)nanopore_nucmer_results_tig00000864 = nanopore_nucmer_results %>%
filter(col.4 > 1000) %>%
filter(col.3 == "tig00000864") %>%
arrange(desc(perID), desc(col.4) ) %>%
mutate(id = row_number(),
start = col.1,
end = col.2,
name = col.3,
strand = col.5,
chrom = col.0)
ggplotly(ggplot(nanopore_nucmer_results_tig00000864) +
geom_rect(aes(xmin = actualStart, xmax = actualEnd,
ymin = id, ymax = id + 1,
fill = col.0,
chrom = chrom,
start = start,
end = end,
mapLength = mapLength,
name = name,
actualStart = actualStart,
actualEnd = actualEnd,
actualLength = actualLength,
strand = strand)) +
scale_fill_tableau("Tableau 20") +
sofonias_theme)bwa mem -t 40 -M assembly/PfSD01nano.fasta /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R1.fastq.gz /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R2.fastq.gz | samtools sort -@ 40 -o bams/PfSD01-to-PfSD01nano.sorted.bam
samtools index bams/PfSD01-to-PfSD01nano.sorted.bamPlotting coverage against the Pacbio assembled PfHB3(Otto et al. 2018)
Plotting across the whole of chromosome 11 in 1kb windows stepping very 500 bases
PfHB3_11_windows_size1000_step500 = readr::read_tsv("data/PfHB3_11_windows_size1000_step500.tab.txt") %>%
mutate(medianPerBaseCov = median(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = perBaseCoverage/medianPerBaseCov) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNorm / 0.5)*0.5)
ggplot(PfHB3_11_windows_size1000_step500) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage,
fill = factor(perBaseCoverageNormRounded)
)) + scale_fill_tableau(name = "Normalized Coverage") + sofonias_theme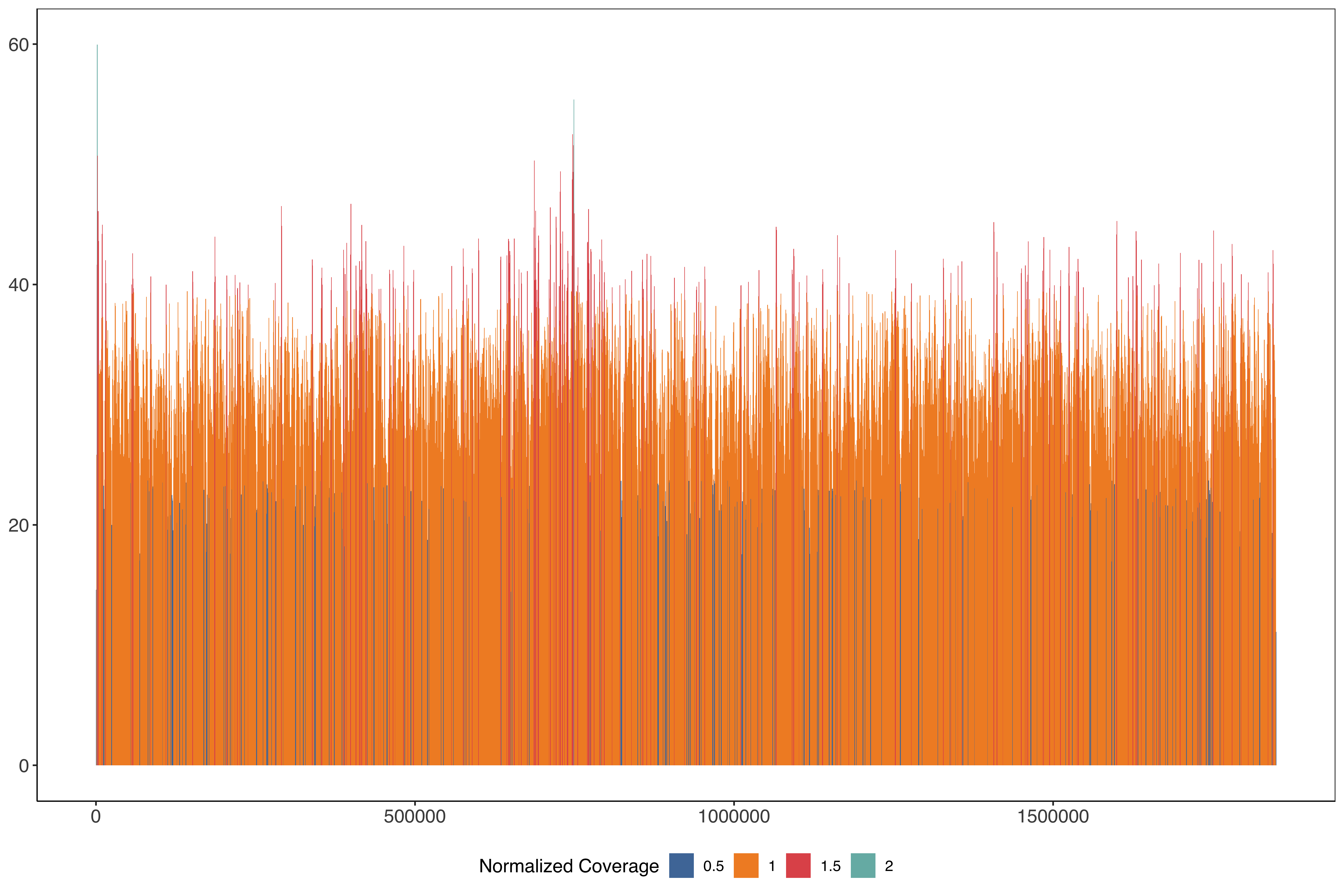
PfHB3_11_windows_size1000_step500_filt = PfHB3_11_windows_size1000_step500 %>%
filter(start >= min(all11$start))
allRepeats_filt_starts_ends_tares_chrom11 = allRepeats_filt_starts_ends_tares %>%
filter(grepl("_11", X1)) %>%
filter(X2 >= min(all11$start))
all11_hb3 = all11 %>% filter(strain == "PfHB3")
PfHB3_chrom11_plot = ggplot(PfHB3_11_windows_size1000_step500_filt) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage
)) + geom_rect(
data = all11_hb3,
aes(
xmin = start,
xmax = end,
ymin = -25,
ymax = 0,
fill = description
),
color = "black"
)
if(nrow(allRepeats_filt_starts_ends_tares_chrom11 %>% filter(strain == "PfHB3")) > 0){
PfHB3_chrom11_plot = PfHB3_chrom11_plot+
geom_rect(
data = allRepeats_filt_starts_ends_tares_chrom11 %>% filter(strain == "PfHB3"),
aes(
xmin = start,
xmax = end,
ymin = -25/2,
ymax = 0,
fill = repeatType,
color = repeatType
)
)
}
PfHB3_chrom11_plot = PfHB3_chrom11_plot+
sofonias_theme +
scale_fill_manual(values = descriptionColors[names(descriptionColors) %in% c(all11$description, "SB3", "TARE1")])+
labs(title = "Illumina Coverage PfHB3 Pacbio assembly Chr11")
ggplotly(PfHB3_chrom11_plot)PfHB3_chrom11_plot = PfHB3_chrom11_plot +
scale_fill_manual(
"Genes",
values = geneAnnotationsColors,
breaks = names(rawGeneAnnotationsColors),
labels = names(rawGeneAnnotationsColors),
guide = guide_legend(
override.aes = list(
color = rawGeneAnnotationsColors[unique(all11_hb3$description)],
fill = rawGeneAnnotationsColors[unique(all11_hb3$description)],
alpha = 1,
shape = 22,
size = 5
),
nrow = 4,
order = 1
)
) +
scale_color_manual(
"Genomic\nElements",
# values = geneAnnotationsColors,
values = c("black", "black"),
breaks = names(genomicElementsColors),
labels = names(genomicElementsColors),
guide = guide_legend(
override.aes = list(
color = genomicElementsColors,
fill = genomicElementsColors,
alpha = 1,
shape = 22,
size = 5
),
ncol = 1,
order = 2
)
)
pdf("PfHB3_illumina_against_pacbio_chrom11_plot.pdf", width = 17.5, height = 8, useDingbats = F)
print(PfHB3_chrom11_plot)
dev.off()quartz_off_screen
2 Plotting across the whole of chromosome 13 in 1kb windows stepping very 500 bases
PfHB3_13_windows_size1000_step500 = readr::read_tsv("data/PfHB3_13_windows_size1000_step500.tab.txt")%>%
mutate(medianPerBaseCov = median(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = perBaseCoverage/medianPerBaseCov) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNorm / 0.5)*0.5)
ggplot(PfHB3_13_windows_size1000_step500) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage,
fill = factor(perBaseCoverageNormRounded)
)) + scale_fill_tableau(name = "Normalized Coverage") + sofonias_theme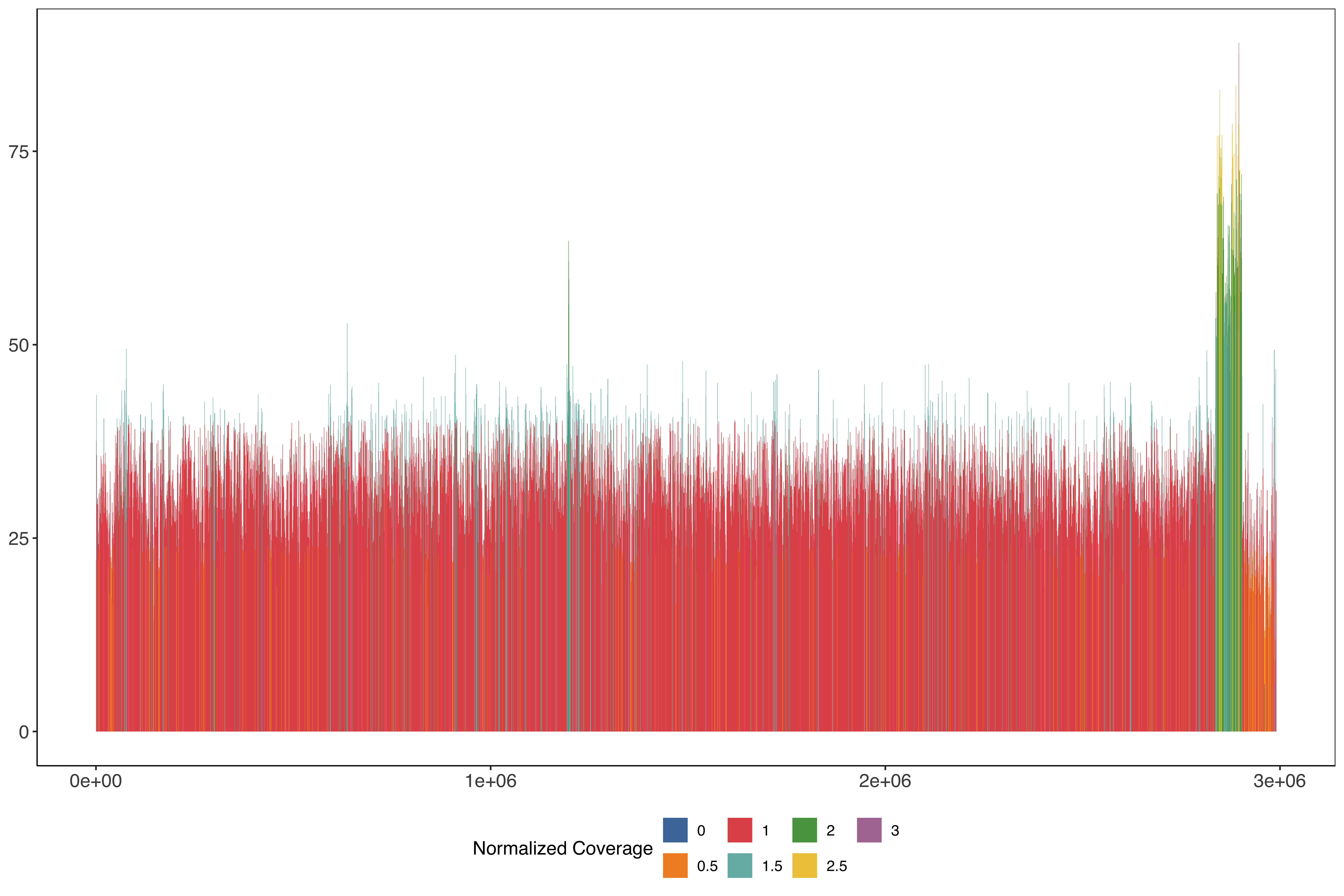
PfHB3_13_windows_size1000_step500_filt = PfHB3_13_windows_size1000_step500 %>%
filter(start >= min(all13$start))
allRepeats_filt_starts_ends_tares_chrom13 = allRepeats_filt_starts_ends_tares %>%
filter(grepl("_13", X1)) %>%
filter(X2 >= min(all13$start))
all13_hb3 = all13 %>% filter(strain == "PfHB3")
PfHB3_chrom13_plot = ggplot(PfHB3_13_windows_size1000_step500_filt) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage
)) + geom_rect(
data =all13_hb3,
aes(
xmin = start,
xmax = end,
ymin = -25,
ymax = 0,
fill = description
),
color = "black"
)
if(nrow(allRepeats_filt_starts_ends_tares_chrom13 %>% filter(strain == "PfHB3")) > 0){
PfHB3_chrom13_plot = PfHB3_chrom13_plot+
geom_rect(
data = allRepeats_filt_starts_ends_tares_chrom13 %>% filter(strain == "PfHB3"),
aes(
xmin = start,
xmax = end,
ymin = -25/2,
ymax = 0,
fill = repeatType,
color = repeatType
)
)
}
PfHB3_chrom13_plot = PfHB3_chrom13_plot+
sofonias_theme +
scale_fill_manual(values = descriptionColors[names(descriptionColors) %in% c(all13$description, "SB3", "TARE1")])+
scale_color_manual(values = descriptionColors[names(descriptionColors) %in% c(all13$description, "SB3", "TARE1")])+
labs(title = "Illumina Coverage PfHB3 Pacbio assembly Chr13")
ggplotly(PfHB3_chrom13_plot)allRepeats_filt_starts_ends_tares_chrom13_PfHB3 = allRepeats_filt_starts_ends_tares_chrom13 %>% filter(strain == "PfHB3")
PfHB3_chrom13_plot = PfHB3_chrom13_plot +
scale_fill_manual(
"Genes",
values = geneAnnotationsColors,
breaks = names(rawGeneAnnotationsColors),
labels = names(rawGeneAnnotationsColors),
guide = guide_legend(
override.aes = list(
color = rawGeneAnnotationsColors[unique(all13_hb3$description)],
fill = rawGeneAnnotationsColors[unique(all13_hb3$description)],
alpha = 1,
shape = 22,
size = 5
),
nrow = 5,
order = 1
)
) +
scale_color_manual(
"Genomic\nElements",
# values = geneAnnotationsColors,
values = c("black"),
breaks = names(genomicElementsColors[names(genomicElementsColors) %in% allRepeats_filt_starts_ends_tares_chrom13_PfHB3$repeatType]),
labels = names(genomicElementsColors[names(genomicElementsColors) %in% allRepeats_filt_starts_ends_tares_chrom13_PfHB3$repeatType]),
guide = guide_legend(
override.aes = list(
color = genomicElementsColors[names(genomicElementsColors) %in% allRepeats_filt_starts_ends_tares_chrom13_PfHB3$repeatType],
fill = genomicElementsColors[names(genomicElementsColors) %in% allRepeats_filt_starts_ends_tares_chrom13_PfHB3$repeatType],
alpha = 1,
shape = 22,
size = 5
),
ncol = 1,
order = 2
)
)
pdf("PfHB3_illumina_against_pacbio_chrom13_plot.pdf", width = 17.5, height = 8, useDingbats = F)
print(PfHB3_chrom13_plot)
dev.off()quartz_off_screen
2 rRNA_28s = readr::read_tsv("../../HB3_new_assembly/extract_28s_rRNA_regions/Pf3D7_11_v3-1928933-1933138-for/beds/reOriented_PfHB3_nanopore_11_13_region.bed", col_names = F) %>%
rename(col.0 = X1,
col.1 = X2,
col.2 = X3,
col.3 = X4,
col.4 = X5,
col.5 = X6) %>%
mutate(ID = "PfHB3_rRNA-28s", feature = "rRNA", product = "rRNA", product_mod = "rRNA")
annotations = readr::read_tsv("../../HB3_new_assembly/sharedRegionBlast/reOriented_PfHB3_nanopore_11_13/renamed_ids_near_ends_withInfo.tab.txt")
annotations = annotations %>%
mutate(product_mod = gsub("term=", "", product)) %>%
#rename(chrom = col.0, start = col.1, end = col.2, name = col.3, length = col.4, strand = col.5) %>%
# mutate(gene = ifelse(grepl("PF3D7_0831800", product_mod), "HRP II", "other")) %>%
# mutate(gene = ifelse(grepl("PF3D7_1372200", product_mod), "HRP III", gene)) %>%
mutate(product_mod = gsub("unknownfunction", "unknown function", product_mod)) %>%
mutate(product_mod = gsub(" \\(SURFIN", "\\(SURFIN", product_mod)) %>%
mutate(product_mod = ifelse(grepl("Plasmodium exported protein", product_mod), "Plasmodium exported protein (PHIST)", product_mod)) %>%
mutate(product_mod = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==product_mod, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", product_mod)) %>%
mutate(product_mod = ifelse("membrane associated erythrocyte binding-like protein"==product_mod, "merozoite adhesive erythrocytic binding protein", product_mod)) %>%
mutate(product_mod = ifelse("membrane associated erythrocyte binding-likeprotein"==product_mod, "merozoite adhesive erythrocytic binding protein", product_mod)) %>%
mutate(product_mod = ifelse("membrane associated histidine-rich protein 1"==product_mod, "membrane associated histidine-rich protein", product_mod)) %>%
mutate(product_mod = gsub(",putative", ", putative", product_mod)) %>%
mutate(product_mod = gsub(",pseudogene", ", pseudogene", product_mod))%>%
mutate(product_mod = gsub("unknownfunction", "unknown function", product_mod))%>%
mutate(product_mod = gsub("conserved protein, unknown function", "conserved Plasmodium protein, unknown function", product_mod)) %>%
mutate(product_mod = gsub(" \\(SURFIN", "\\(SURFIN", product_mod)) %>%
mutate(product_mod = ifelse(grepl("Plasmodium exported protein", product_mod), "Plasmodium exported protein (PHIST)", product_mod)) %>%
mutate(product_mod = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==product_mod, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", product_mod)) %>%
mutate(product_mod = ifelse("membrane associated histidine-rich protein 1"==product_mod, "membrane associated histidine-rich protein", product_mod))%>%
mutate(product_mod = gsub(",pseudogene", ", pseudogene", product_mod))%>%
mutate(product_mod = gsub("unknownfunction", "unknown function", product_mod))%>%
mutate(product_mod = gsub(" \\(SURFIN", "\\(SURFIN", product_mod)) %>%
mutate(product_mod = ifelse(grepl("Plasmodium exported protein", product_mod), "Plasmodium exported protein (PHIST)", product_mod)) %>%
mutate(product_mod = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==product_mod, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", product_mod)) %>%
mutate(product_mod = ifelse(grepl("sporozoite and liver stage tryptophan-rich protein, putative", product_mod), "tryptophan/threonine-rich antigen", product_mod))%>%
mutate(product_mod = ifelse(grepl("CRA domain-containing protein, putative", product_mod), "conserved Plasmodium protein, unknown function", product_mod))%>%
mutate(product_mod = gsub(",pseudogene", ", pseudogene", product_mod)) %>%
mutate(product_mod = gsub("surfaceantigen", "surface antigen", product_mod)) %>%
mutate(product_mod = gsub("Tetratricopeptide repeat, putative", "tetratricopeptide repeat protein, putative", product_mod)) %>%
mutate(product_mod = gsub("transmembraneprotein", "transmembrane protein", product_mod)) %>%
mutate(product_mod = ifelse(grepl("PfEMP1", product_mod) & grepl("pseudogene", product_mod), "erythrocyte membrane protein 1 (PfEMP1), pseudogene", product_mod)) %>%
mutate(product_mod = gsub("PIR protein", "stevor", product_mod)) %>%
mutate(product_mod = gsub("erythrocyte membrane protein 1-like", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", product_mod)) %>%
mutate(product_mod = gsub("acidic terminal segments, variant surface antigen of PfEMP1, putative", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", product_mod))%>%
mutate(product_mod = ifelse(grepl("CoA binding protein", product_mod, ignore.case = T), "acyl-CoA binding protein", product_mod)) %>%
mutate(product_mod = ifelse(grepl("transfer RNA", product_mod) | grepl("tRNA", product_mod), "tRNA", product_mod))%>%
mutate(product_mod = ifelse(grepl("cytoadherence", product_mod), "CLAG", product_mod))%>%
mutate(product_mod = ifelse(grepl("surface-associated interspersed protein", product_mod), "SURFIN", product_mod))%>%
mutate(product_mod = ifelse(grepl("SURFIN", product_mod), "SURFIN", product_mod))%>%
mutate(product_mod = ifelse(grepl("stevor-like", product_mod), "stevor, pseudogene", product_mod)) %>%
mutate(product_mod = ifelse(grepl("exported protein family", product_mod), "exported protein family", product_mod)) %>%
mutate(product_mod = ifelse(grepl("rRNA", feature), "rRNA", product_mod)) %>%
mutate(product_mod = ifelse(grepl("serine/threonine protein kinase", product_mod), "serine/threonine protein kinase, FIKK family", product_mod)) %>%
mutate(product_mod = ifelse(grepl("hypothetical protein", product_mod), "hypothetical protein, conserved", product_mod)) %>%
mutate(product_mod = ifelse(grepl("conserved Plasmodium protein, unknown function", product_mod), "hypothetical protein, conserved", product_mod)) %>%
mutate(product_mod = ifelse(grepl("Rifin/stevor family, putative", product_mod), "stevor", product_mod)) %>%
mutate(product_mod = ifelse(grepl("erythrocyte membrane protein 1 (PfEMP1), pseudogene", product_mod), "erythrocyte membrane protein 1 (PfEMP1)", product_mod)) %>%
mutate(product_mod = ifelse(grepl("erythrocyte membrane protein 1", product_mod), "erythrocyte membrane protein 1 (PfEMP1)", product_mod))%>%
mutate(product_mod = ifelse(grepl("Plasmodium RESA N-terminal, putative", product_mod), "ring-infected erythrocyte surface antigen", product_mod)) %>%
left_join(rRNA_28s %>%
select(col.0, col.1, col.2) %>%
rename(`28rRNA-start` = col.1,
`28rRNA-end` = col.2)) %>%
filter(!(col.1 >= `28rRNA-start` & col.1 <= `28rRNA-end`)) %>%
bind_rows(rRNA_28s)
backtraceAmount = 50000
annotations_filt = annotations %>%
filter((col.0 == "PfHB3_11" & col.1 >=1849728 -backtraceAmount) |
(col.0 == "PfHB3_13" & col.1 >=2827490 -backtraceAmount)) %>%
filter(!("hypothetical protein, conserved" == product_mod &
((col.0 == "PfHB3_11" & col.1 >=1944764) |
(col.0 == "PfHB3_13" & col.1 >=2859471))))
annotations_filt_blank = tibble(
col.0 = c("PfHB3_11", "PfHB3_13"),
col.1 = c(1849728 -backtraceAmount, 2827490 -backtraceAmount))
HB3chromsLens = readr::read_tsv("../../HB3_new_assembly/fakeGenomes/chroms_reOriented_PfHB3_nanopore_11_13.txt", col_names =c( "chrom", "len"))
HB3chromsLens_mod = HB3chromsLens %>%
rename(col.0 = chrom, col.2 = len) %>%
left_join(annotations_filt_blank %>%
select(col.0, col.1) %>%
rename(start = col.1)) %>%
mutate(len = col.2 -start)
annotations_filt_blank = annotations_filt_blank %>%
left_join(HB3chromsLens_mod %>%
select(col.0, len)) %>%
mutate(col.2 = col.1 + max(len)) %>%
mutate(newLen = col.2 - col.1)
rawGeneAnnotationsColors = scheme$hex(length(unique(c(annotations_filt$product_mod))))
names(rawGeneAnnotationsColors) = unique(c(annotations_filt$product_mod))
geneAnnotationsColors = c(rawGeneAnnotationsColors, genomicElementsColors)
tares = readr::read_tsv("../../HB3_new_assembly/repeats_filt_starts_ends_tares.tsv") %>%
mutate(chrom = X1,
start = X2,
end = X3)
tares_chrom11_end = tares %>%
filter(X1 == "PfHB3nano_11") %>%
filter(!is.na(endPos))
tares_chrom13_end = tares %>%
filter(X1 == "PfHB3nano_13") %>%
filter(!is.na(endPos))
tares_chrom11_chrom13_ends = bind_rows(
tares_chrom11_end,
tares_chrom13_end
)
tares_chrom11_chrom13_ends = tares_chrom11_chrom13_ends %>%
mutate(start = X2,
end = X3)
annotations_filt = annotations_filt %>%
mutate(col.0 = gsub("PfHB3", "PfHB3nano", col.0)) %>%
mutate(chrom = col.0,
start = col.1,
end = col.2)
annotations_filt_chrom11 = annotations_filt %>%
filter(col.0 == "PfHB3nano_11")
annotations_filt_chrom13 = annotations_filt %>%
filter(col.0 == "PfHB3nano_13")Plotting coverage against the new HB3 assembled with nanopore
PfHB3_nano_11_windows_size1000_step500 = readr::read_tsv("data/PfHB3nano_11_windows_size1000_step500.tab.txt")%>%
mutate(medianPerBaseCov = median(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = perBaseCoverage/medianPerBaseCov) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNorm / 0.5)*0.5)
ggplot(PfHB3_nano_11_windows_size1000_step500) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage,
fill = factor(perBaseCoverageNormRounded)
)) + scale_fill_tableau(name = "Normalized Coverage") + sofonias_theme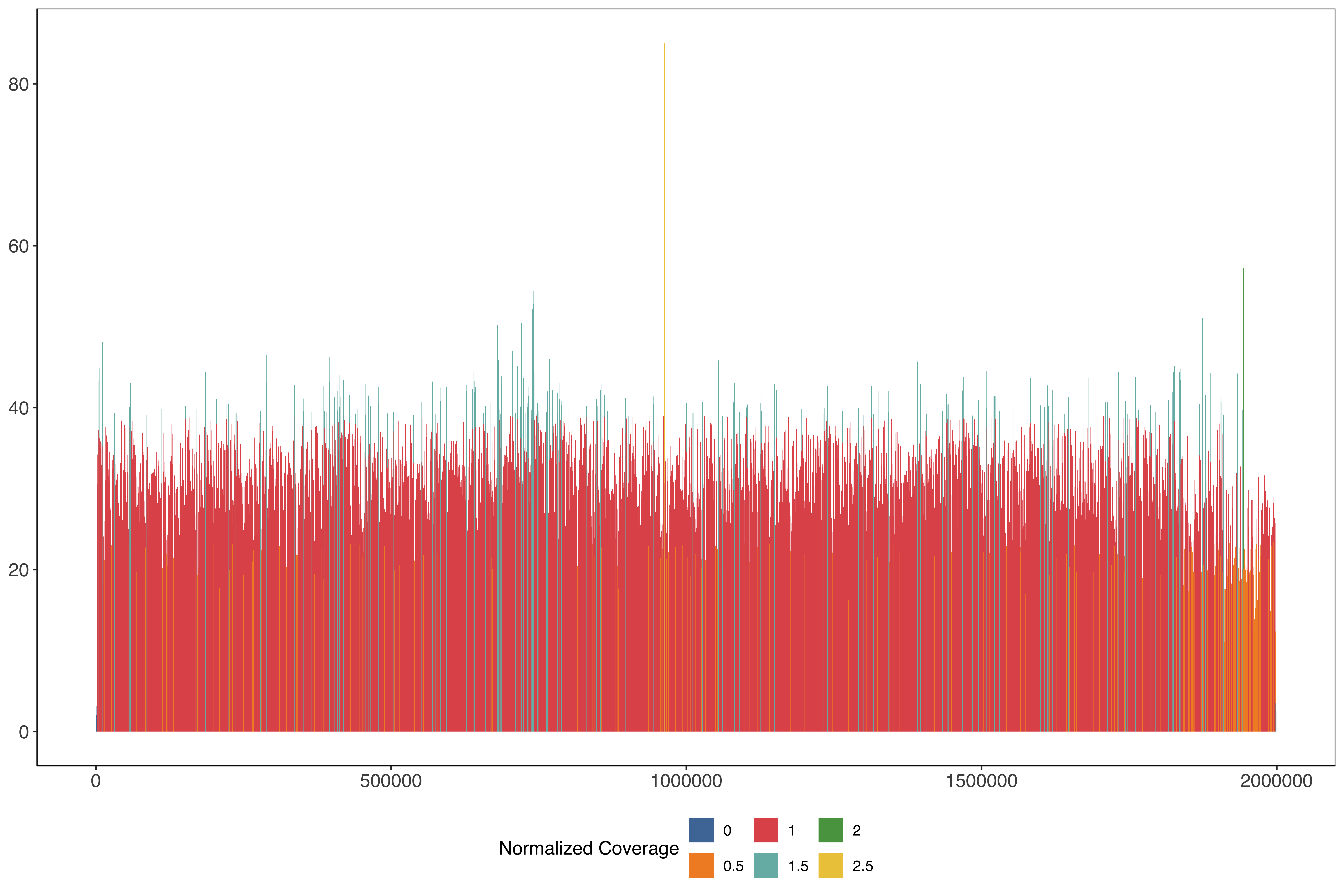
PfHB3_nano_11_windows_size1000_step500_filt = PfHB3_nano_11_windows_size1000_step500 %>%
filter(start >= min(annotations_filt_chrom11$start))
PfHB3_chrom11_plot = ggplot(PfHB3_nano_11_windows_size1000_step500_filt) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage
)) + geom_rect(
data = annotations_filt_chrom11,
aes(
xmin = start,
xmax = end,
ymin = -25,
ymax = 0,
fill = product_mod
),
color = "black"
)
if(nrow(tares_chrom11_end) > 0){
PfHB3_chrom11_plot = PfHB3_chrom11_plot+
geom_rect(
data = tares_chrom11_end,
aes(
xmin = start,
xmax = end,
ymin = -25/2,
ymax = 0,
fill = repeatType,
color = repeatType
)
)
}
PfHB3_chrom11_plot = PfHB3_chrom11_plot+
sofonias_theme +
scale_fill_manual(values = geneAnnotationsColors[names(geneAnnotationsColors) %in% c(annotations_filt_chrom11$product_mod, "SB3", "TARE1")])+
scale_color_manual(values = c("black", "black")) +
labs(title = "Illumina Coverage PfHB3 Nano assembly Chr11")
ggplotly(PfHB3_chrom11_plot)PfHB3_chrom11_plot = PfHB3_chrom11_plot +
scale_fill_manual(
"Genes",
values = geneAnnotationsColors,
breaks = names(rawGeneAnnotationsColors),
labels = names(rawGeneAnnotationsColors),
guide = guide_legend(
override.aes = list(
color = rawGeneAnnotationsColors[unique(annotations_filt_chrom11$product_mod)],
fill = rawGeneAnnotationsColors[unique(annotations_filt_chrom11$product_mod)],
alpha = 1,
shape = 22,
size = 5
),
nrow = 4,
order = 1
)
) +
scale_color_manual(
"Genomic\nElements",
# values = geneAnnotationsColors,
values = c("black", "black"),
breaks = names(genomicElementsColors),
labels = names(genomicElementsColors),
guide = guide_legend(
override.aes = list(
color = genomicElementsColors,
fill = genomicElementsColors,
alpha = 1,
shape = 22,
size = 5
),
ncol = 1,
order = 2
)
)
pdf("PfHB3_illumina_against_nano_chrom11_plot.pdf", width = 17.5, height = 8, useDingbats = F)
print(PfHB3_chrom11_plot)
dev.off()quartz_off_screen
2 PfHB3_nano_13_windows_size1000_step500 = readr::read_tsv("data/PfHB3nano_13_windows_size1000_step500.tab.txt")%>%
mutate(medianPerBaseCov = median(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = perBaseCoverage/medianPerBaseCov) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNorm / 0.5)*0.5)
ggplot(PfHB3_nano_13_windows_size1000_step500) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage,
fill = factor(perBaseCoverageNormRounded)
)) + scale_fill_tableau(name = "Normalized Coverage") + sofonias_theme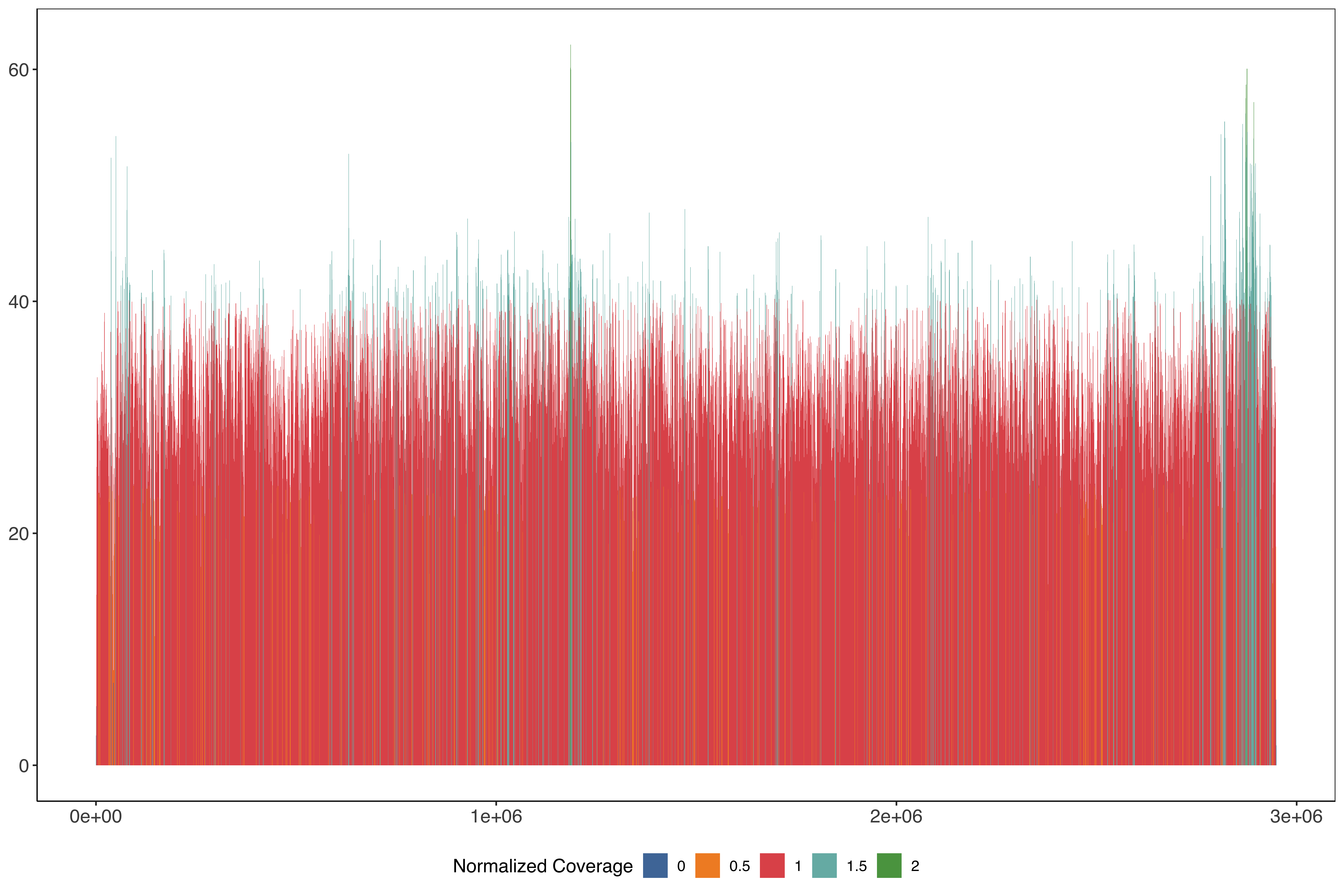
PfHB3_nano_13_windows_size1000_step500_filt = PfHB3_nano_13_windows_size1000_step500 %>%
filter(start >= min(annotations_filt_chrom13$start))
PfHB3_chrom13_plot = ggplot(PfHB3_nano_13_windows_size1000_step500_filt) +
geom_rect(aes(
xmin = start,
xmax = end - 1,
ymin = 0,
ymax = perBaseCoverage
)) + geom_rect(
data = annotations_filt_chrom13,
aes(
xmin = start,
xmax = end,
ymin = -25,
ymax = 0,
fill = product_mod
),
color = "black"
)
if(nrow(tares_chrom13_end) > 0){
PfHB3_chrom13_plot = PfHB3_chrom13_plot+
geom_rect(
data = tares_chrom13_end,
aes(
xmin = start,
xmax = end,
ymin = -25/2,
ymax = 0,
fill = repeatType,
color = repeatType
)
)
}
PfHB3_chrom13_plot = PfHB3_chrom13_plot+
sofonias_theme +
scale_fill_manual(values = geneAnnotationsColors[names(geneAnnotationsColors) %in% c(annotations_filt_chrom13$product_mod, "SB3", "TARE1")])+
scale_color_manual(values = c("black", "black")) +
labs(title = "Illumina Coverage PfHB3 Nano assembly Chr13")
ggplotly(PfHB3_chrom13_plot)PfHB3_chrom13_plot = PfHB3_chrom13_plot +
scale_fill_manual(
"Genes",
values = geneAnnotationsColors,
breaks = names(rawGeneAnnotationsColors),
labels = names(rawGeneAnnotationsColors),
guide = guide_legend(
override.aes = list(
color = rawGeneAnnotationsColors[unique(annotations_filt_chrom13$product_mod)],
fill = rawGeneAnnotationsColors[unique(annotations_filt_chrom13$product_mod)],
alpha = 1,
shape = 22,
size = 5
),
nrow = 4,
order = 1
)
) +
scale_color_manual(
"Genomic\nElements",
# values = geneAnnotationsColors,
values = c("black", "black"),
breaks = names(genomicElementsColors),
labels = names(genomicElementsColors),
guide = guide_legend(
override.aes = list(
color = genomicElementsColors,
fill = genomicElementsColors,
alpha = 1,
shape = 22,
size = 5
),
ncol = 1,
order = 2
)
)
pdf("PfHB3_illumina_against_nano_chrom13_plot.pdf", width = 17.5, height = 8, useDingbats = F)
print(PfHB3_chrom13_plot)
dev.off()quartz_off_screen
2