Processing Samples for chr13 TARE1 presence
Set up
Downloads
Reading in data
Code
allMetaSamplesByBio = readr::read_tsv("../../meta/metadata/metaByBioSample.tab.txt")
allMetaSamples = readr::read_tsv("../../meta/metadata/meta.tab.txt")
allMetaDeletionCalls = readr::read_tsv("../metaSelected.tab.txt")
allMetaDeletionCalls_possiblyHRP2Deleted = allMetaDeletionCalls %>%
filter(possiblyHRP2Deleted)
allMetaDeletionCalls_possiblyChr11Deleted = allMetaDeletionCalls %>%
filter(possiblyChr11Deleted)
allMetaDeletionCalls_Hrp3pattern2 = readr::read_tsv("../allMetaDeletionCalls_Hrp3pattern2.tab.txt")
realmccoilCoiCalls = readr::read_tsv("../wgs_variants/THEREALMcCOIL/categorical_method/real_mccoil_COI_calls.tsv")
realmccoilCoiCalls_poly = realmccoilCoiCalls %>%
filter(random_median != 1 | topHE_median != 1) %>%
left_join(allMetaSamples %>%
select(sample, BiologicalSample))Creating a region to investigate on chromosome 13
Code
finalHRPII_HRPIII_windows = readr::read_tsv("../../windowAnalysis/windows/finalHRPII_HRPIII_windows_withTuned.bed", col_names = F) %>%
mutate(genomicID = paste0(X1, "-", X2, "-", X3)) %>%
mutate(chrom = X1, len = X3 -X2) %>%
rename(name = X4)
finalHRPII_HRPIII_windows_investTelemeraseHealing = finalHRPII_HRPIII_windows %>%
filter("Pf3D7_13_v3" == X1, X2 >= 2813887)
write_tsv(finalHRPII_HRPIII_windows_investTelemeraseHealing %>%
select(1:7), "finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_largeRegions.bed", col_names = F)Code
finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full = finalHRPII_HRPIII_windows %>%
filter("Pf3D7_13_v3" == X1, X2 >= 2817793)
finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out = finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full %>%
group_by(X1) %>% summarise(X2 = min(X2),
X3 = max(X3)) %>%
mutate(name = paste0(X1, "-", X2, "-", X3)) %>%
mutate(len = X3 - X2,
strand = "+")
write_tsv(finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out %>%
select(1:6), "finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out.bed", col_names = F)Create sub-windows within this large window to investigate
Code
elucidator createWindowsInRegions --bed finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out.bed --step 1000 --windowSize 1000 --minLen 50 | elucidator bedGetIntersectingGenesInGff --gff /tank/data/genomes/plasmodium/genomes/pf/info/gff/Pf3D7.gff --extraAttributes description --bed STDIN --overWrite --out finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000.bed
elucidator createWindowsInRegions --bed finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out.bed --step 100 --windowSize 100 --minLen 50 | elucidator bedGetIntersectingGenesInGff --gff /tank/data/genomes/plasmodium/genomes/pf/info/gff/Pf3D7.gff --extraAttributes description --bed STDIN --overWrite --out finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step100_windowSize100.bed
rsync -raPh finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000.bed hathawan@calderon.barrel-of-knowledge.info:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22
rsync -raPh finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step100_windowSize100.bed hathawan@calderon.barrel-of-knowledge.info:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22
rsync -raPh ../allMetaDeletionCalls_Hrp3pattern2_samples.txt hathawan@calderon.barrel-of-knowledge.info:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22Finding regions with TARE1 sequence
Code
nohup elucidator runMultipleCommands --cmdFile runRegionCmds.txt --additionalFields "{DIRNAME}:finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_NoTrim_keep;{SAMPLE}:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000.bed\";{NCPUS}:4 --keepTemporaryFiles --qualCheck 5 --keepImproperMates" --replaceFields --numThreads 11 &
cd finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_NoTrim_keep
PathWeaver runProcessClustersOnRecon --overWriteDir --dout popClustering --pat _finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_NoTrim_keep --numThreads 5 --groupingsFile /tank/data/plasmodium/falciparum/pfdata/metadata/meta.tab.txt --samples /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt
# 5`-3` "TT[CT]AGGGTT[CT]AGGG"
# 3`-5` "CCCT[AG]AACCCT[AG]AA"
elucidator countPWExtractedReadsWithPattern --seqPat "TT[CT]AGGGTT[CT]AGGG","CCCT[AG]AACCCT[AG]AA" --pat _finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_NoTrim_keep --bedFnp /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000.bed --minReadCounts 1 --numThreads 10 --overWrite --out finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_countTares.txt --samples /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txtCode
nohup elucidator runMultipleCommands --cmdFile runRegionCmds.txt --additionalFields "{DIRNAME}:finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step100_windowSize100_NoTrim_keep;{SAMPLE}:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step100_windowSize100.bed\"" --replaceFields --numThreads 11 &
cd finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step100_windowSize100_NoTrim_keep
PathWeaver runProcessClustersOnRecon --overWriteDir --dout popClustering --pat _finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step100_windowSize100_NoTrim_keep --numThreads 5 --groupingsFile /tank/data/plasmodium/falciparum/pfdata/metadata/meta.tab.txt --samples /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt Code
finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_countTares = readr::read_tsv("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_NoTrim_keep/finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_countTares.txt")
finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_countTares_sum = finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_countTares %>%
group_by(sample, region, seqPat) %>%
summarise(count = sum(count))
hrp3Pat2_samplesWithTare1 = finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_countTares_sum %>%
ungroup() %>%
select(sample, region) %>%
unique() %>%
rename(breakwithinRegion = region)Plotting coverage
Code
cov = readr::read_tsv("../../meta/allCov_summaryStats.tab.txt.gz")
allReports = readr::read_tsv("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_NoTrim_keep/popClustering/reports/allBasicInfo.tab.txt.gz") %>%
mutate(inGene = !is.na(extraField0)) %>%
left_join(cov) %>%
mutate(medianCov = median(perBaseCoverage),
meanCov = mean(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = ifelse(inGene, perBaseCoverage/medianPerBaseCov_inGenes, perBaseCoverage/medianPerBaseCov_notInGenes)) %>%
mutate(perBaseCoverageNormRounded = ifelse(perBaseCoverageNorm >0.10 & perBaseCoverageNorm <= 1, 1, perBaseCoverageNorm)) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNormRounded)) %>%
filter(sample %!in% realmccoilCoiCalls_poly$sample)
allReports_sp = allReports %>%
select(sample, name, perBaseCoverageNormRounded) %>%
spread(name, perBaseCoverageNormRounded)
allReports_sp_mat = as.matrix(allReports_sp[,2:ncol(allReports_sp)])
rownames(allReports_sp_mat) = allReports_sp$sample
allReports_sp_mat[allReports_sp_mat >2] = 2
annotationTextSize = 20
topAnno = allReports %>%
filter(sample == .$sample[1]) %>%
select(name, extraField0) %>%
rename(`Gene Description` = extraField0) %>%
mutate(`Gene Description` = gsub(".*description=", "", `Gene Description`))%>%
mutate(`Gene Description` = gsub("\\].*", "", `Gene Description`)) %>%
mutate(`Gene Description` = ifelse(grepl("PHIST", `Gene Description`), gsub("\\).*", "", gsub(".*PHIST", "PHIST", `Gene Description`)), `Gene Description`))
topAnnoDf = topAnno %>%
select(-name) %>%
as.data.frame()
topAnnoColors = createColorListFromDf(topAnnoDf)
allReports_sp_mat_sampleOrder = hclust(dist(allReports_sp_mat))$order
allReports_sp_mat_sampleOrderDf = tibble(
sample = rownames(allReports_sp_mat)[allReports_sp_mat_sampleOrder],
order = 1:nrow(allReports_sp_mat)
#order = allReports_sp_mat_sampleOrder
)
metaReSelected = allMetaDeletionCalls[match(rownames(allReports_sp_mat), allMetaDeletionCalls$sample), ] %>%
left_join(allReports_sp_mat_sampleOrderDf)
rowAnno = tibble(metaReSelected[,c("sample","country", "order")]) %>%
mutate(`TARE1 Present On Chr13` = sample %in% hrp3Pat2_samplesWithTare1$sample) %>%
#mutate(`Transposition With 5` = sample %in% samplesWithTransitionToChr5$sample) %>%
#select(-order)
#arrange(`TARE1 Present On Chr13`, `Transposition With 5`, order) %>%
arrange(order) %>%
select(-order)
metaReSelected = metaReSelected[match(rowAnno$sample, metaReSelected$sample),]
allReports_sp_mat = allReports_sp_mat[match(rowAnno$sample, rownames(allReports_sp_mat)), ]
rowAnnoDf = rowAnno %>%
select(-sample) %>%
as.data.frame()
rowAnnoColors = createColorListFromDf(rowAnnoDf)
sideAnno = rowAnnotation(
df = rowAnnoDf,
col = rowAnnoColors,
gp = gpar(col = "grey10"),
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
gap = unit(0.1, "cm")
)
topAnno = HeatmapAnnotation(
df = topAnnoDf,
col = topAnnoColors,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
na_col = "#FFFFFF00"
)
library(circlize)
col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3)))
allReports_sp_mat_nolab = allReports_sp_mat
colnames(allReports_sp_mat_nolab) = NULL
allReports_sp_mat_hm = Heatmap(
allReports_sp_mat_nolab,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno,
right_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
title = "coverage",
at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
cell_fun = function(j, i, x, y, width, height, fill) {
grid.rect(
x = x,
y = y,
width = width,
height = height * .9,
gp = gpar(fill = fill, col = NA)
)
},
rect_gp = gpar(type = "none"),
column_title = "Chr13[2,817kb:2,844kb]",
column_title_gp = gpar(fontsize = 20, fontface = "italic")
)
allReports_sp_mat_hm_noCluster = Heatmap(
allReports_sp_mat_nolab,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno,
right_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
title = "coverage",
at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
cell_fun = function(j, i, x, y, width, height, fill) {
grid.rect(x = x, y = y, width = width, height = height *.9,
gp = gpar(fill = fill, col = NA))
},
rect_gp = gpar(type = "none"),
column_title = "Chr13[2,817kb:2,844kb]",
column_title_gp = gpar(fontsize = 20, fontface = "italic")
)Final Plot
Code
draw(allReports_sp_mat_hm, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", padding = unit(c(40, 2, 2, 2), "mm")) # , legend_border = T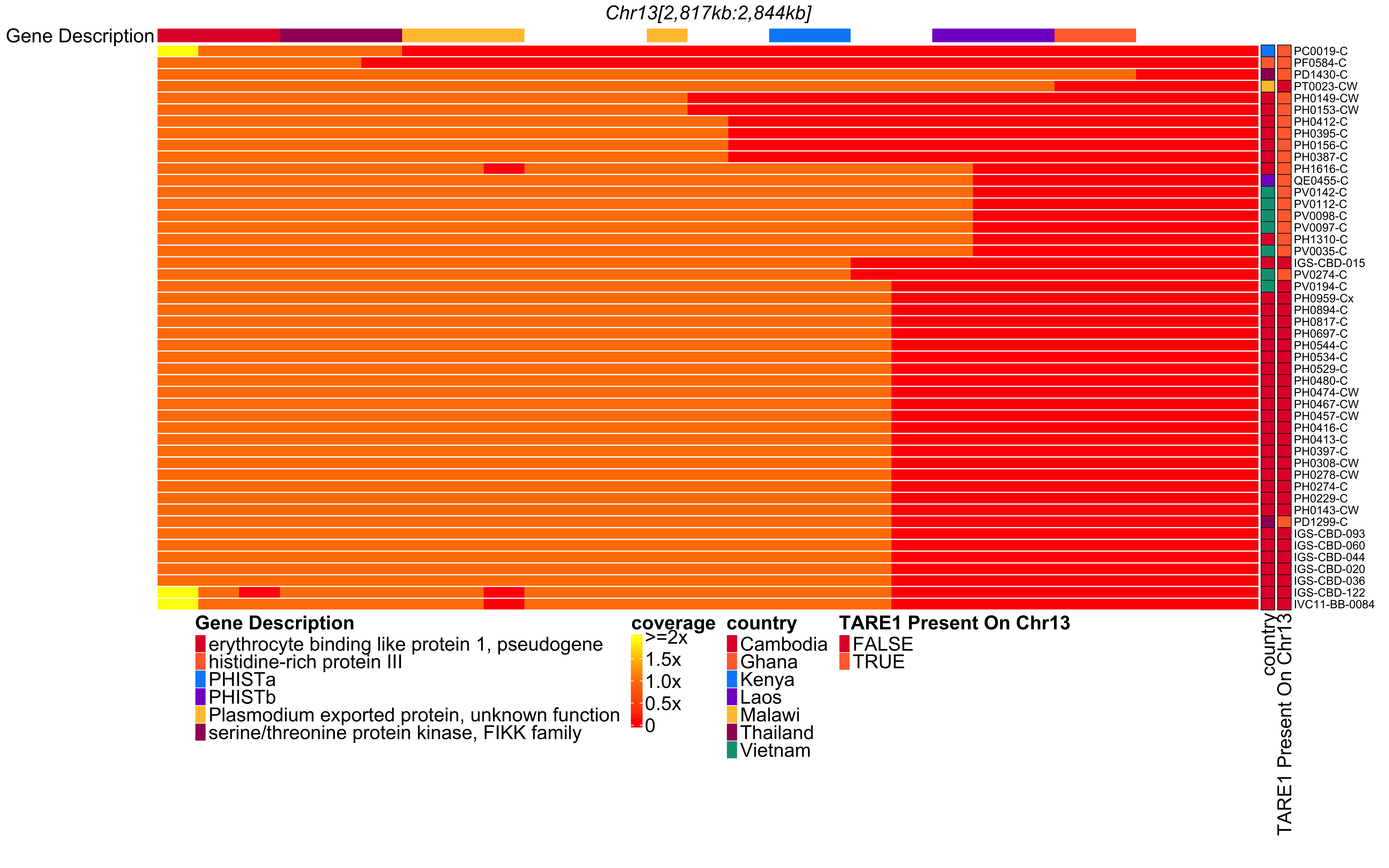
Code
cairo_pdf("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000.pdf", width = 20, height = 12.5)
draw(allReports_sp_mat_hm, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", padding = unit(c(40, 2, 2, 2), "mm")) # , legend_border = T
dev.off()quartz_off_screen
2 Plotting presence of TARE1 to match up with coverage
Code
hrp3Pat2_samplesWithTare1_filler = expand_grid(
sample = rownames(allReports_sp_mat),
breakwithinRegion = colnames(allReports_sp_mat)
) %>%
mutate(marker = 0)
hrp3Pat2_samplesWithTare1_sel = hrp3Pat2_samplesWithTare1 %>%
select(sample, breakwithinRegion) %>%
mutate(marker = 1) %>%
bind_rows(hrp3Pat2_samplesWithTare1_filler) %>%
group_by(sample, breakwithinRegion) %>%
summarise(marker = sum(marker))
hrp3Pat2_samplesWithTare1_sel_sp = hrp3Pat2_samplesWithTare1_sel %>%
group_by() %>%
spread(breakwithinRegion, marker)
hrp3Pat2_samplesWithTare1_sel_sp_mat = as.matrix(hrp3Pat2_samplesWithTare1_sel_sp[,2:ncol(hrp3Pat2_samplesWithTare1_sel_sp)])
rownames(hrp3Pat2_samplesWithTare1_sel_sp_mat) = hrp3Pat2_samplesWithTare1_sel_sp$sample
hrp3Pat2_samplesWithTare1_sel_sp_mat = hrp3Pat2_samplesWithTare1_sel_sp_mat[match(rownames(allReports_sp_mat), rownames(hrp3Pat2_samplesWithTare1_sel_sp_mat)),]
hrp3Pat2_samplesWithTare1_sel_sp_mat_noLab = hrp3Pat2_samplesWithTare1_sel_sp_mat
colnames(hrp3Pat2_samplesWithTare1_sel_sp_mat_noLab) = NULL
allReports_sp_mat_hm_tare1Present = Heatmap(
hrp3Pat2_samplesWithTare1_sel_sp_mat_noLab,
cluster_columns = F,
cluster_rows = F,
col = colorRamp2(c(0, 1), c("#FFFFFF00", "green")),
name = "coverage",
top_annotation = topAnno,
right_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
title = "coverage",
at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
cell_fun = function(j, i, x, y, width, height, fill) {
grid.rect(
x = x,
y = y,
width = width,
height = height * .9,
gp = gpar(fill = fill, col = NA)
)
},
rect_gp = gpar(type = "none")
)Code
cairo_pdf("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr13_full_out_step1000_windowSize1000_tare1_Present.pdf", width = 20, height = 12.5, onefile = T)
draw(allReports_sp_mat_hm_noCluster, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", padding = unit(c(20, 2, 2, 2), "mm"))
draw(allReports_sp_mat_hm_tare1Present, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", padding = unit(c(20, 2, 2, 2), "mm"))
dev.off()quartz_off_screen
2 hrp3Pat2 samples With Tare1 on chr13
Code
hrp3Pat2_samplesWithTare1 = hrp3Pat2_samplesWithTare1 %>%
left_join(allMetaDeletionCalls)
create_dt(hrp3Pat2_samplesWithTare1)Code
hrp3Pat2_samplesWithTare1_mod = hrp3Pat2_samplesWithTare1 %>%
separate(breakwithinRegion, into = c("chrom", "start", "end", "strand"), remove = F, convert = T, sep = "-")
hrp3Pat2_samplesWithTare1_mod_filt = hrp3Pat2_samplesWithTare1_mod %>%
group_by(sample) %>%
slice_max(end)
pf3d7_chrom13_coreEnding = 2853482
hrp3Pat2_samplesWithTare1_mod_filt =hrp3Pat2_samplesWithTare1_mod_filt %>%
mutate(pf3d7_chrom13_core_deletion = pf3d7_chrom13_coreEnding - end)
write_tsv(hrp3Pat2_samplesWithTare1, "hrp3Pat2_samplesWithTare1.tsv")Amount of “core” genome deleted with telomere healing on chromosome 13
Code
skimr::skim(hrp3Pat2_samplesWithTare1_mod_filt %>% as_data_frame())| Name | hrp3Pat2_samplesWithTare1… |
| Number of rows | 20 |
| Number of columns | 24 |
| _______________________ | |
| Column type frequency: | |
| character | 14 |
| Date | 1 |
| logical | 5 |
| numeric | 4 |
| ________________________ | |
| Group variables | None |
Variable type: character
| skim_variable | n_missing | complete_rate | min | max | empty | n_unique | whitespace |
|---|---|---|---|---|---|---|---|
| sample | 0 | 1 | 8 | 9 | 0 | 20 | 0 |
| breakwithinRegion | 0 | 1 | 31 | 31 | 0 | 7 | 0 |
| chrom | 0 | 1 | 11 | 11 | 0 | 1 | 0 |
| strand | 0 | 1 | 3 | 3 | 0 | 1 | 0 |
| country | 0 | 1 | 4 | 8 | 0 | 6 | 0 |
| site | 0 | 1 | 6 | 10 | 0 | 8 | 0 |
| ExperimentSample | 0 | 1 | 8 | 8 | 0 | 20 | 0 |
| BiologicalSample | 0 | 1 | 8 | 8 | 0 | 20 | 0 |
| region | 0 | 1 | 11 | 22 | 0 | 4 | 0 |
| subRegion | 0 | 1 | 11 | 11 | 0 | 3 | 0 |
| secondaryRegion | 0 | 1 | 4 | 6 | 0 | 2 | 0 |
| hrpCall | 0 | 1 | 15 | 15 | 0 | 1 | 0 |
| isolate | 0 | 1 | 5 | 5 | 0 | 1 | 0 |
| hrp3DeletionPattern | 0 | 1 | 9 | 9 | 0 | 1 | 0 |
Variable type: Date
| skim_variable | n_missing | complete_rate | min | max | median | n_unique |
|---|---|---|---|---|---|---|
| collection_date | 19 | 0.05 | 2012-09-01 | 2012-09-01 | 2012-09-01 | 1 |
Variable type: logical
| skim_variable | n_missing | complete_rate | mean | count |
|---|---|---|---|---|
| PreferredSample | 0 | 1 | 1 | TRU: 20 |
| IsFieldSample | 0 | 1 | 1 | TRU: 20 |
| possiblyHRP2Deleted | 0 | 1 | 0 | FAL: 20 |
| possiblyHRP3Deleted | 0 | 1 | 1 | TRU: 20 |
| possiblyChr11Deleted | 0 | 1 | 0 | FAL: 20 |
Variable type: numeric
| skim_variable | n_missing | complete_rate | mean | sd | p0 | p25 | p50 | p75 | p100 | hist |
|---|---|---|---|---|---|---|---|---|---|---|
| start | 0 | 1 | 2833493.00 | 4856.90 | 2821793 | 2830793 | 2835793 | 2836793 | 2840793 | ▂▁▅▇▁ |
| end | 0 | 1 | 2834493.00 | 4856.90 | 2822793 | 2831793 | 2836793 | 2837793 | 2841793 | ▂▁▅▇▁ |
| collection_year | 0 | 1 | 2009.55 | 2.67 | 2002 | 2009 | 2010 | 2011 | 2014 | ▁▁▇▇▅ |
| pf3d7_chrom13_core_deletion | 0 | 1 | 18989.00 | 4856.90 | 11689 | 15689 | 16689 | 21689 | 30689 | ▁▇▅▁▂ |
Code
allReports_tare1Present = allReports %>%
filter(sample %in% hrp3Pat2_samplesWithTare1$sample) %>%
filter(perBaseCoverageNormRounded > 0) %>%
group_by(sample) %>%
slice_max(order_by = start)
allReports_tare1Present_sel = allReports_tare1Present %>%
select(sample, name, `#chrom`, start, extraField0) %>%
left_join(allMetaDeletionCalls %>%
select(sample, country, region)) %>%
arrange(start)
create_dt(allReports_tare1Present_sel)Code
allReports_tare1Present_sel_startCounts = allReports_tare1Present_sel %>% group_by(start) %>% count() %>%
arrange(desc(n))
cat( paste0(sum(allReports_tare1Present_sel_startCounts$n), " samples contain evidence of telomere healing on chromosome 13 resulting in the deletion of pfhrp3 as evidenced by the presence of TARE1(Vernick and McCutchan 1988) contiguous with the genomic sequence at the genomic location where coverage drops off. These breaks occur on chromosome 13 at ", paste0(format(allReports_tare1Present_sel_startCounts$start[1:(length(allReports_tare1Present_sel_startCounts$start) - 1)], nsmall=0, big.mark=","), " (n=", allReports_tare1Present_sel_startCounts$n, "), ", collapse = ""), "and ", format(allReports_tare1Present_sel_startCounts$start[length(allReports_tare1Present_sel_startCounts$start)], nsmall=0, big.mark=","), " (n=", allReports_tare1Present_sel_startCounts$n[length(allReports_tare1Present_sel_startCounts$n)], ")", ". "))19 samples contain evidence of telomere healing on chromosome 13 resulting in the deletion of pfhrp3 as evidenced by the presence of TARE1(Vernick and McCutchan 1988) contiguous with the genomic sequence at the genomic location where coverage drops off. These breaks occur on chromosome 13 at 2,836,793 (n=8), 2,830,793 (n=4), 2,829,793 (n=2), 2,821,793 (n=1), 2,822,793 (n=1), 2,833,793 (n=1), 2,834,793 (n=1), 2,836,793 (n=1), and 2,840,793 (n=1).