Getting specific spanning reads for the variability in the shared region for SD01. The read and overall sample quality of SD01 is poor (fragile DNA) so assembly and long read depth is hard to achieve. This plus the fact that basically from this shared region to the end chromosomes 11 and hybrid 13-11 are perfectly copied (a region close to 200kb) it’s hard to assembly at baseline. For this reason to help support the claim that the hybrid genome 13 exist we can link the same amount of variation seen within the shared region between 11 and 13 to try to link variation within this region to reads that span chromosome 13 into this region. If variation specific to only to chromosome 13 reads are also contained within reads that then span from within the shared region into chromosome 11 sequence (either on the hybrid 13-11 or into 11 since from the shared region to the end of the chromosome this sequence is identical within the 3D7 hybrid genome so reads should be able to multi-map to both these regions).
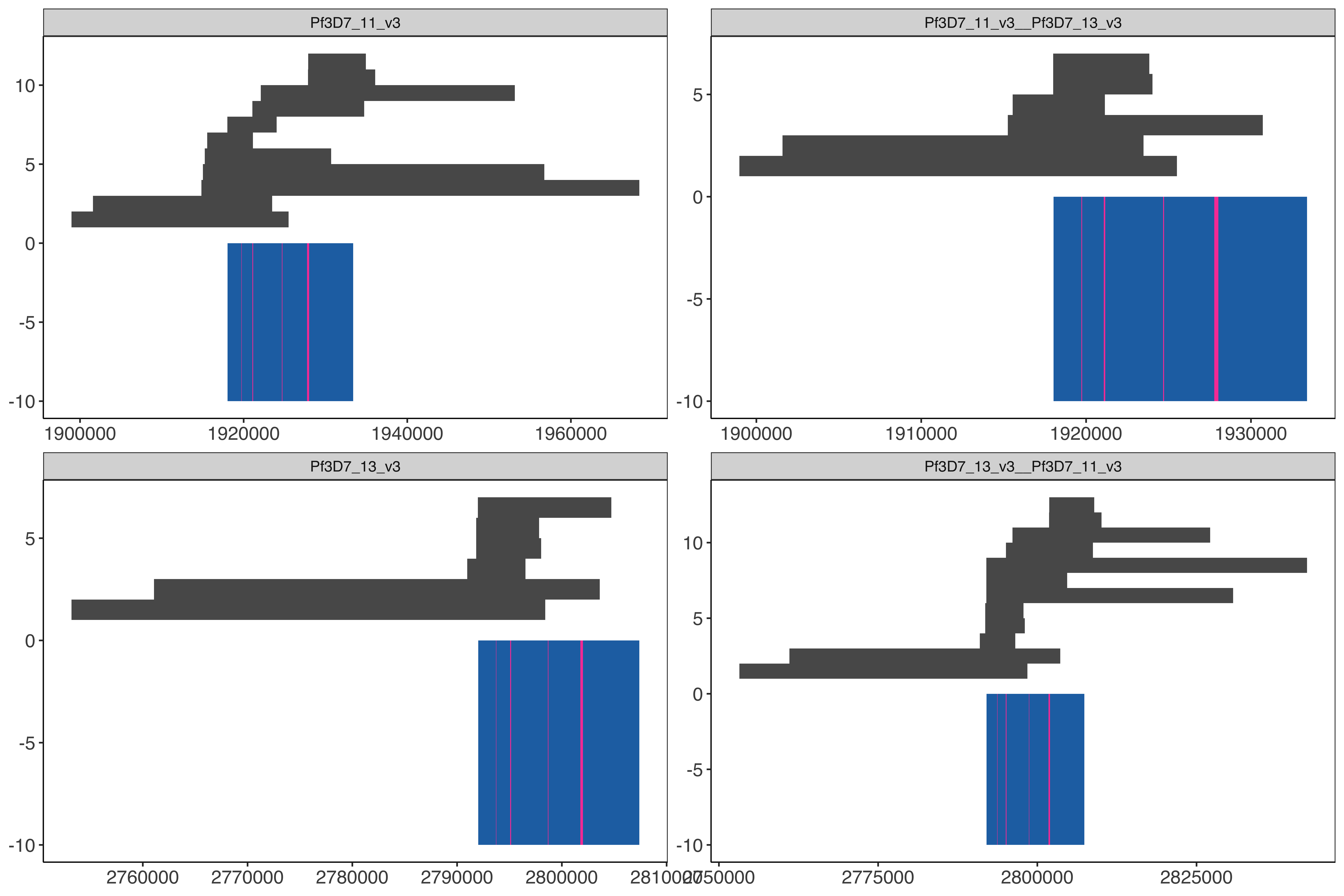
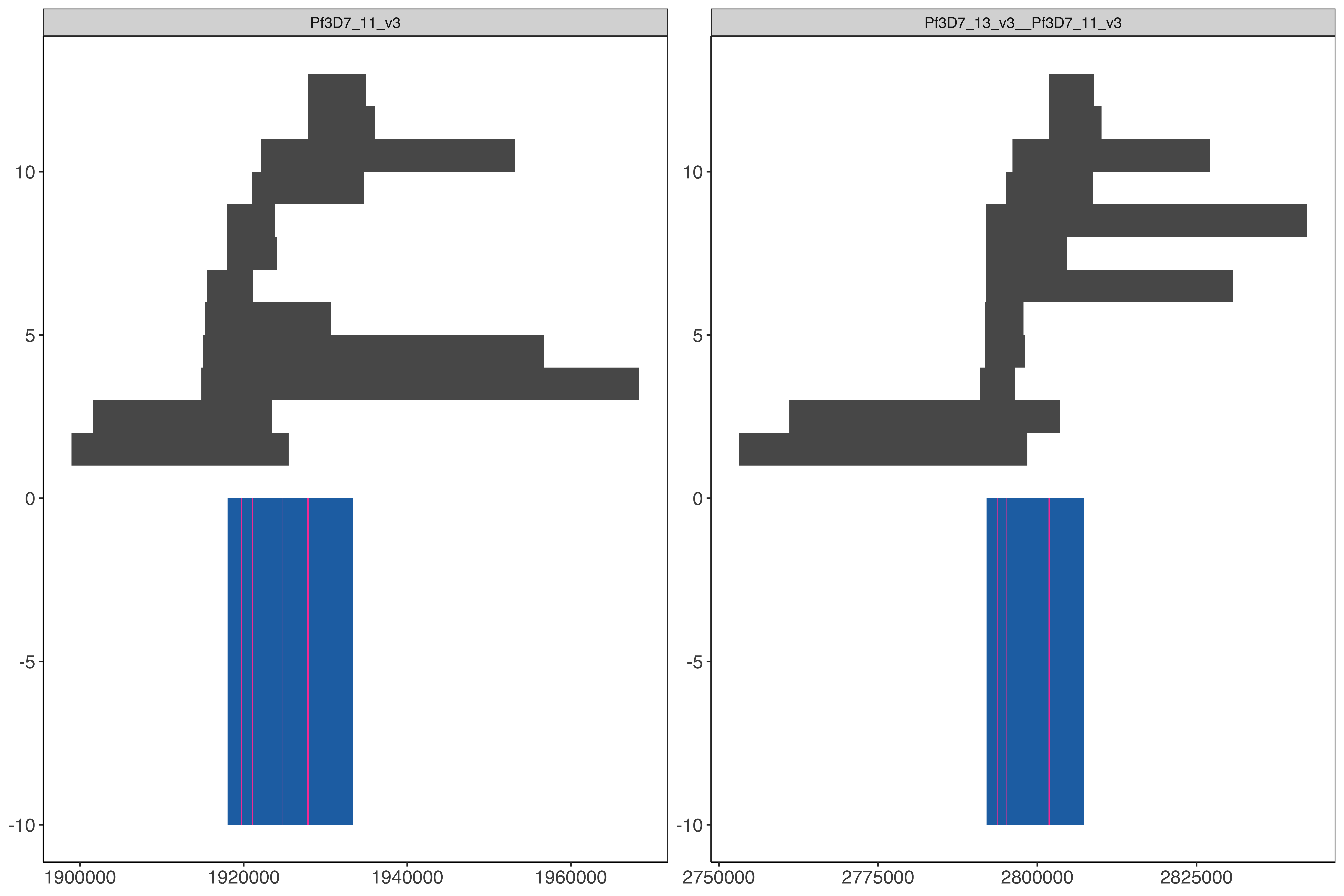
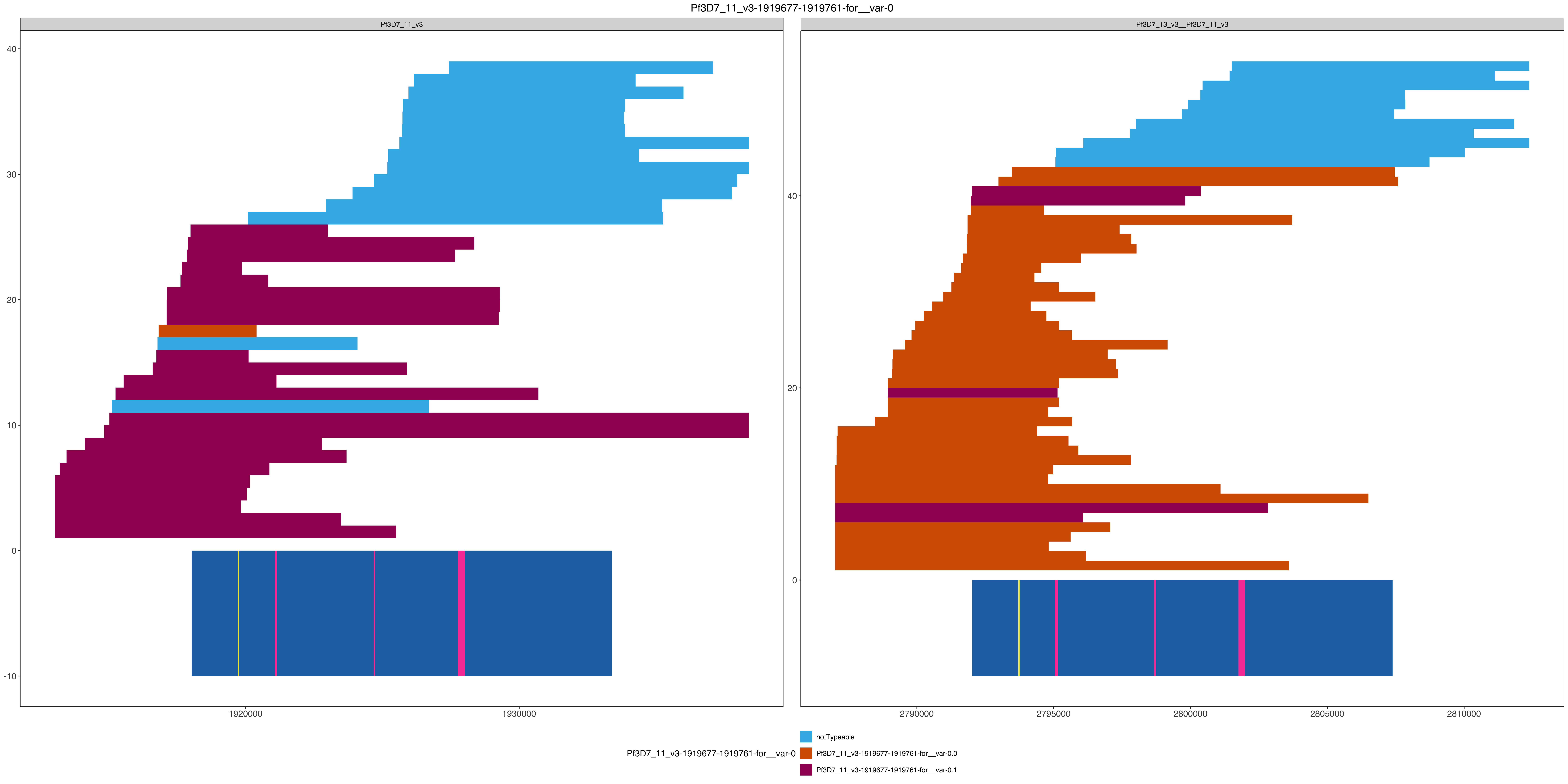
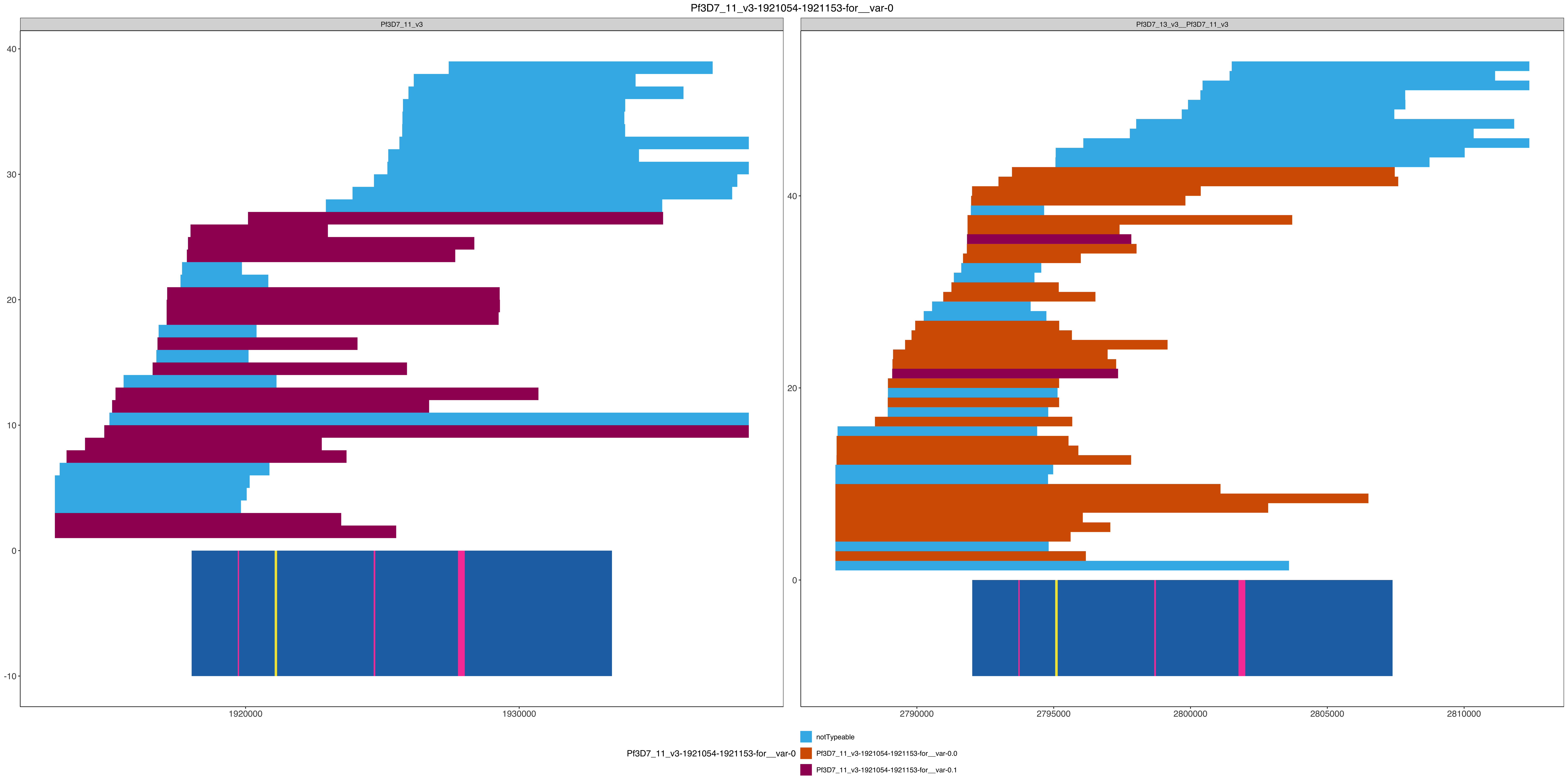
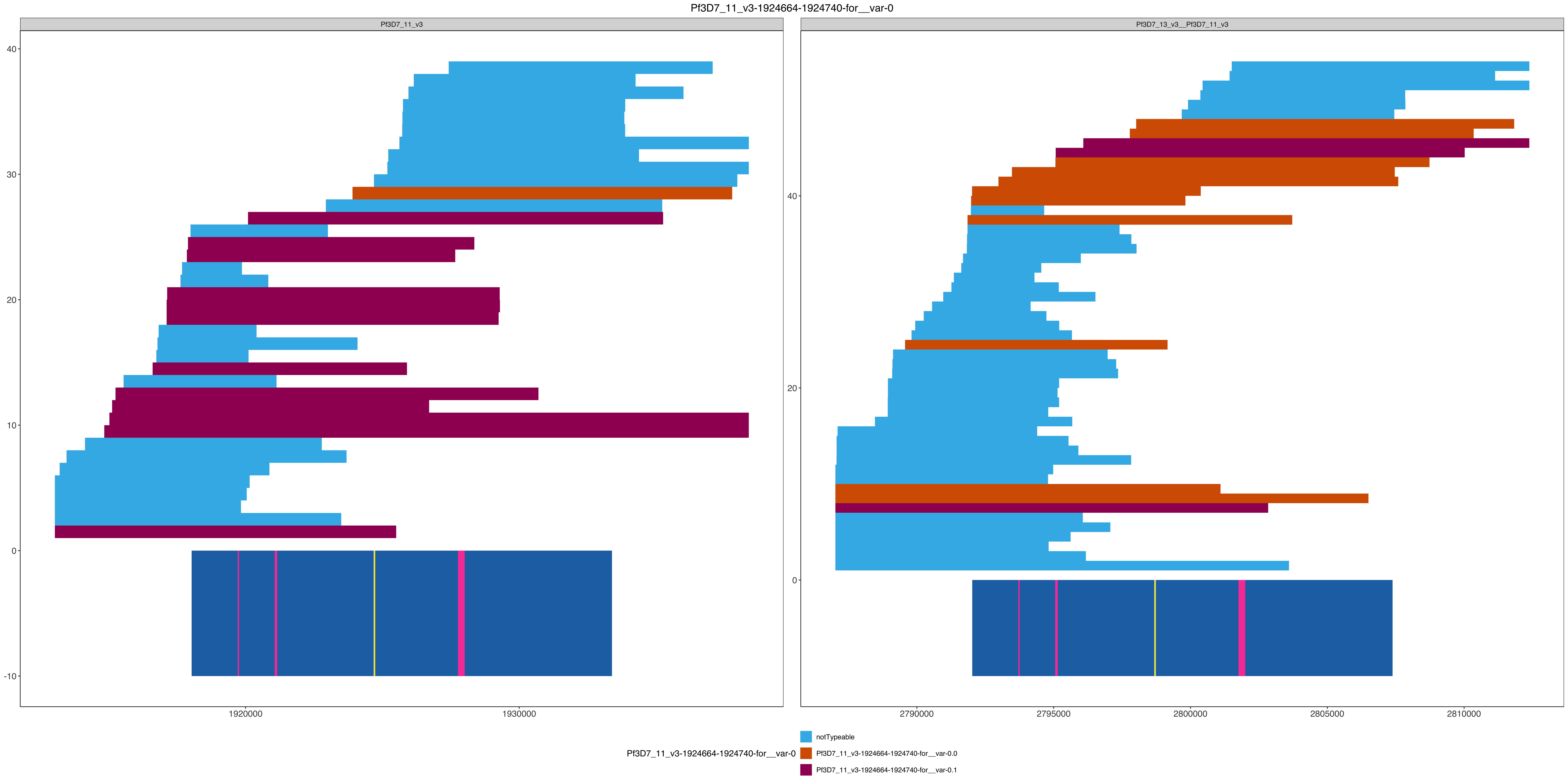
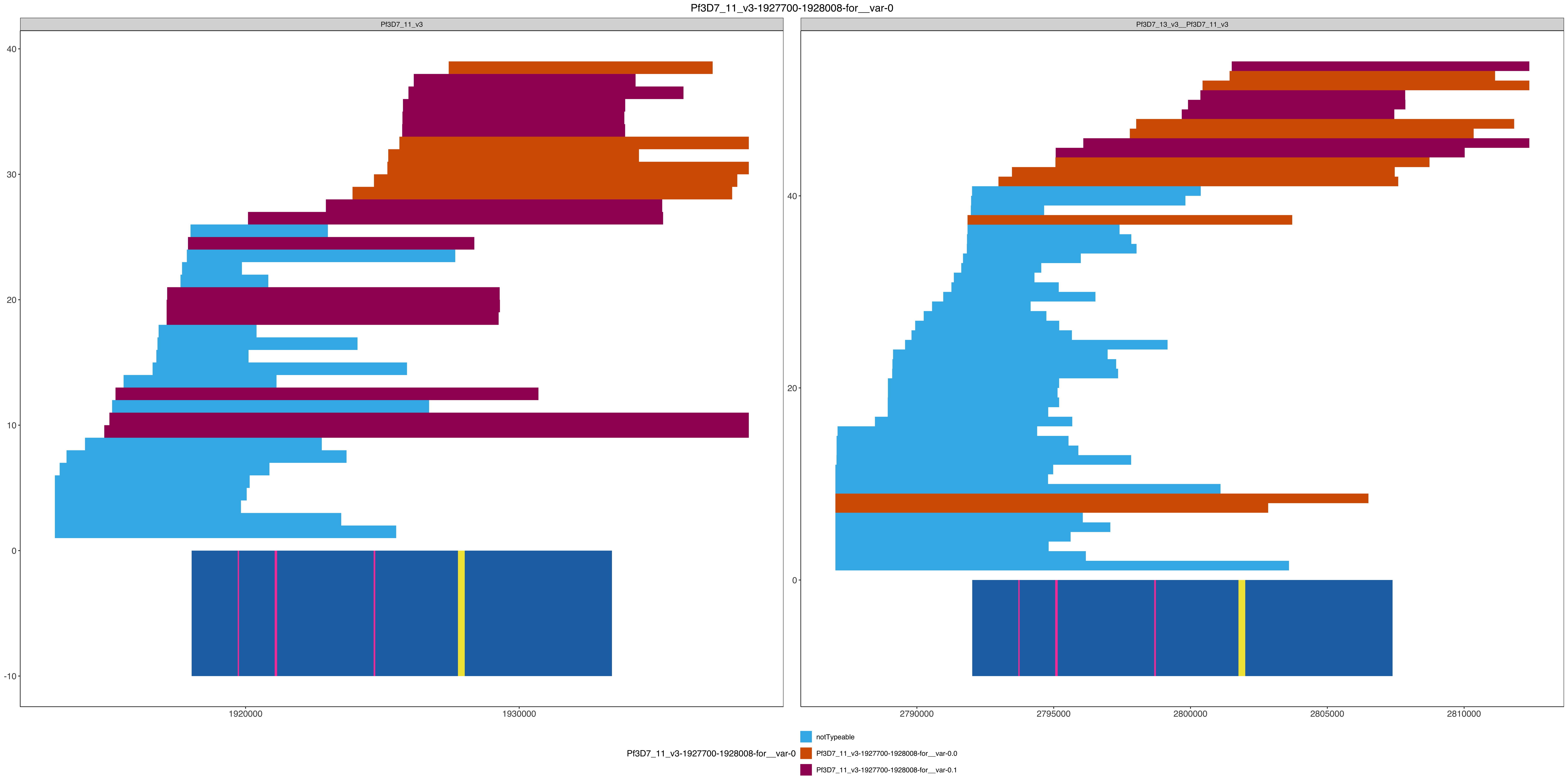
These regions below have variation within the strain SD01 within the 15.3kb shared region between 11 and 13.
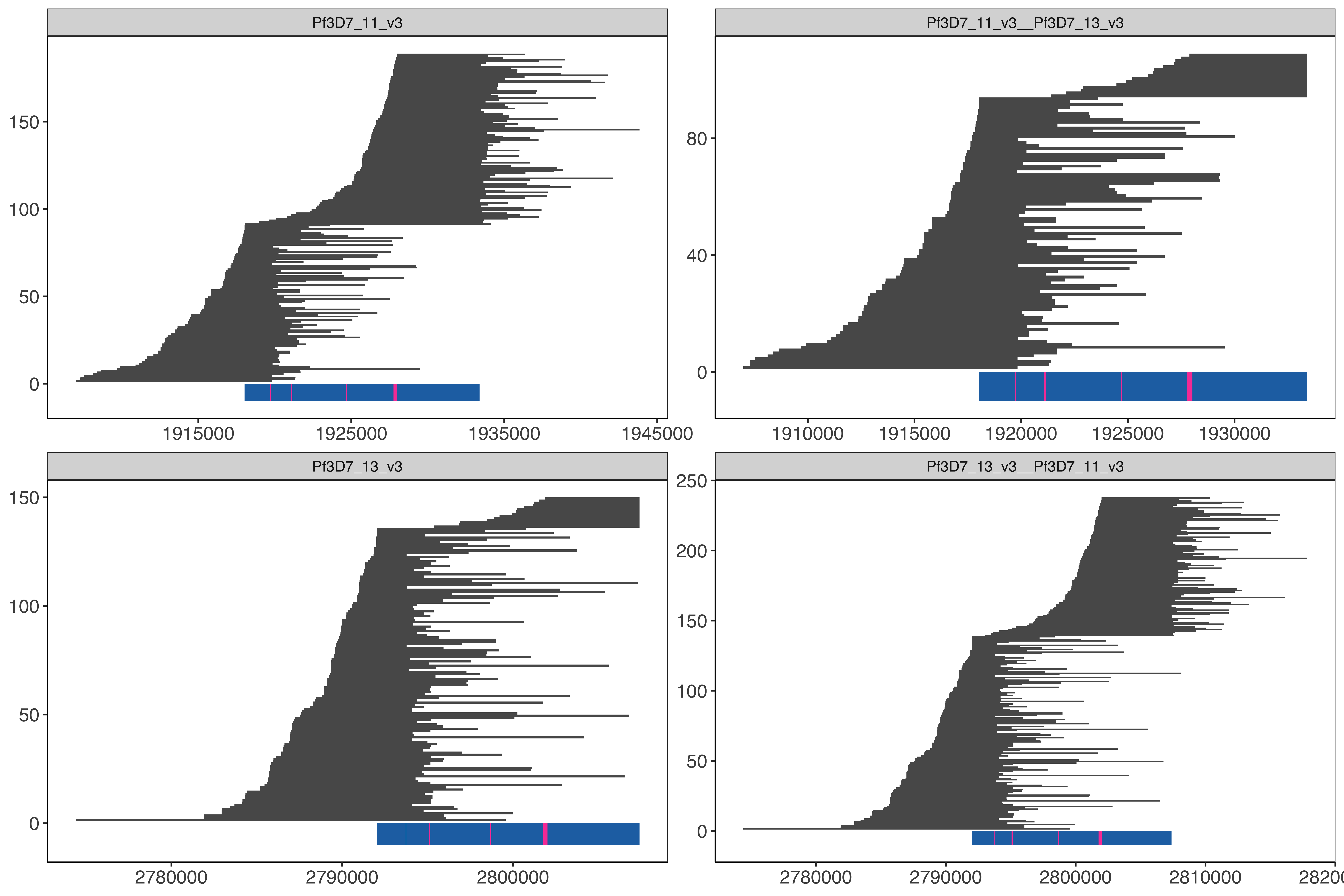
Determine the locations within hybrid genome in order to pull the long reads from the alignments to the hybrid (core 3D7 genome plus hybrid chromosomes chr11-chr13, chr13-chr11) genome
Code
cd ../../../../Windows_Surrounding_HRP2_3_deletions/windowAnalysis/elucidator getFastaWithBed --twoBit /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.2bit --overWrite--bed finalHrpSubwindows_regions_withSD01MultiInShared.bed --out finalHrpSubwindows_regions_withSD01MultiInShared.fasta elucidator determineRegionLastz --identity 100 --coverage 100 --fasta finalHrpSubwindows_regions_withSD01MultiInShared.fasta --individual--genome /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta |sort|uniq> raw_finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid.bed # have to do this cause LASTZ bugs out and doesn't do the matching of Pf3D7_11_v3 1924680 1924740 Pf3D7_11_v3-1924664-1924740-for__var-0 60 + for whatever goodness forsaken reason cut-f1-6 finalHrpSubwindows_regions_withSD01MultiInShared.bed raw_finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid.bed |sort|uniq> finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid.bedelucidator getOverlappingBedRegions --bed finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid.bed --intersectWithBed ../../sharedBetween11_and_13/investigatingChrom11Chrom13/shared_11_13_region_onPureHybrid.bed |cut-f1-6> finalHrpSubwindows_regions_withSD01MultiInShared_inPureHybrid.bed
Get the nanopore/pacbio reads that intersect this region
Add in illumina reads to help with error correcting
Code
elucidator BamGetSpanningReadsForRegionLongReads --bam /tank/data/plasmodium/falciparum/pfdata/bams/PfSD01.sorted.bam --bed /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/finalHrpSubwindows_regions_withSD01MultiInShared.bed --trimToRegion--overWriteDir--dout PfSD01_illumina_finalHrpSubwindows_regions_withSD01MultiInShared_regions --renamemkdir PfSD01_combined_finalHrpSubwindows_regions_withSD01MultiInShared_regionsfor x in PfSD01_illumina_finalHrpSubwindows_regions_withSD01MultiInShared_regions/Pf*gz;doelucidator prependNames --fastqgz${x}--overWrite--prefix illumina_;done;for x in PfSD01_nano_finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid_regions/Pf*gz;doelucidator prependNames --fastqgz${x}--overWrite--prefix nano_;done;for x in PfSD01_pacbio_finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid_regions/Pf*gz;doelucidator prependNames --fastqgz${x}--overWrite--prefix pacbio_;done;
Concatenate reads and add the extraction info
Code
for x in PfSD01_illumina_finalHrpSubwindows_regions_withSD01MultiInShared_regions/prepended_Pf3D7*gz;docat${x} PfSD01_nano_finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid_regions/$(basename${x}) PfSD01_pacbio_finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid_regions/$(basename${x})> PfSD01_combined_finalHrpSubwindows_regions_withSD01MultiInShared_regions/$(basename${x}|sed's/prepended_//g');done;cat PfSD01_nano_finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid_regions/prepended_Pf3D7_11_v3-1927700-1928008-for__var-0.fastq.gz PfSD01_pacbio_finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid_regions/prepended_Pf3D7_11_v3-1927700-1928008-for__var-0.fastq.gz > PfSD01_combined_finalHrpSubwindows_regions_withSD01MultiInShared_regions/Pf3D7_11_v3-1927700-1928008-for__var-0.fastq.gzrsync-raPh PfSD01_illumina_finalHrpSubwindows_regions_withSD01MultiInShared_regions/nameKey.tab.txt PfSD01_combined_finalHrpSubwindows_regions_withSD01MultiInShared_regions/illumina_extraction_nameKey.tab.txtrsync-raPh PfSD01_nano_finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid_regions/nameKey.tab.txt PfSD01_combined_finalHrpSubwindows_regions_withSD01MultiInShared_regions/nano_extraction_nameKey.tab.txtrsync-raPh PfSD01_pacbio_finalHrpSubwindows_regions_withSD01MultiInShared_inHybrid_regions/nameKey.tab.txt PfSD01_combined_finalHrpSubwindows_regions_withSD01MultiInShared_regions/pacbio_extraction_nameKey.tab.txt
Cluster the reads using clustering meant for error prone long reads elucidatorlab kmerClusteringRate to help type the reads.
Code
cd PfSD01_combined_finalHrpSubwindows_regions_withSD01MultiInShared_regions for x in Pf3D7_11_v3-19*gz;doelucidatorlab kmerClusteringRate --qualThres 7,5 --cutOff 0.1 --idCutOff 0.80 --numThreads 15 --lqMismatches 2 --oneBaseIndel 5 --twoBaseIndel 2 --map--recalcConsensus--checkChimeras--overWriteDir--writeInitialClusters--sizeCutOff 10 --fastq${x}--dout clustering_${x%%.fastq.gz}--postCollapseForceOneComp;done;elucidator rBind --contains clusterNames.tab.txt --recursive--delim tab --header--overWrite--out allClusterNames.tab.txt
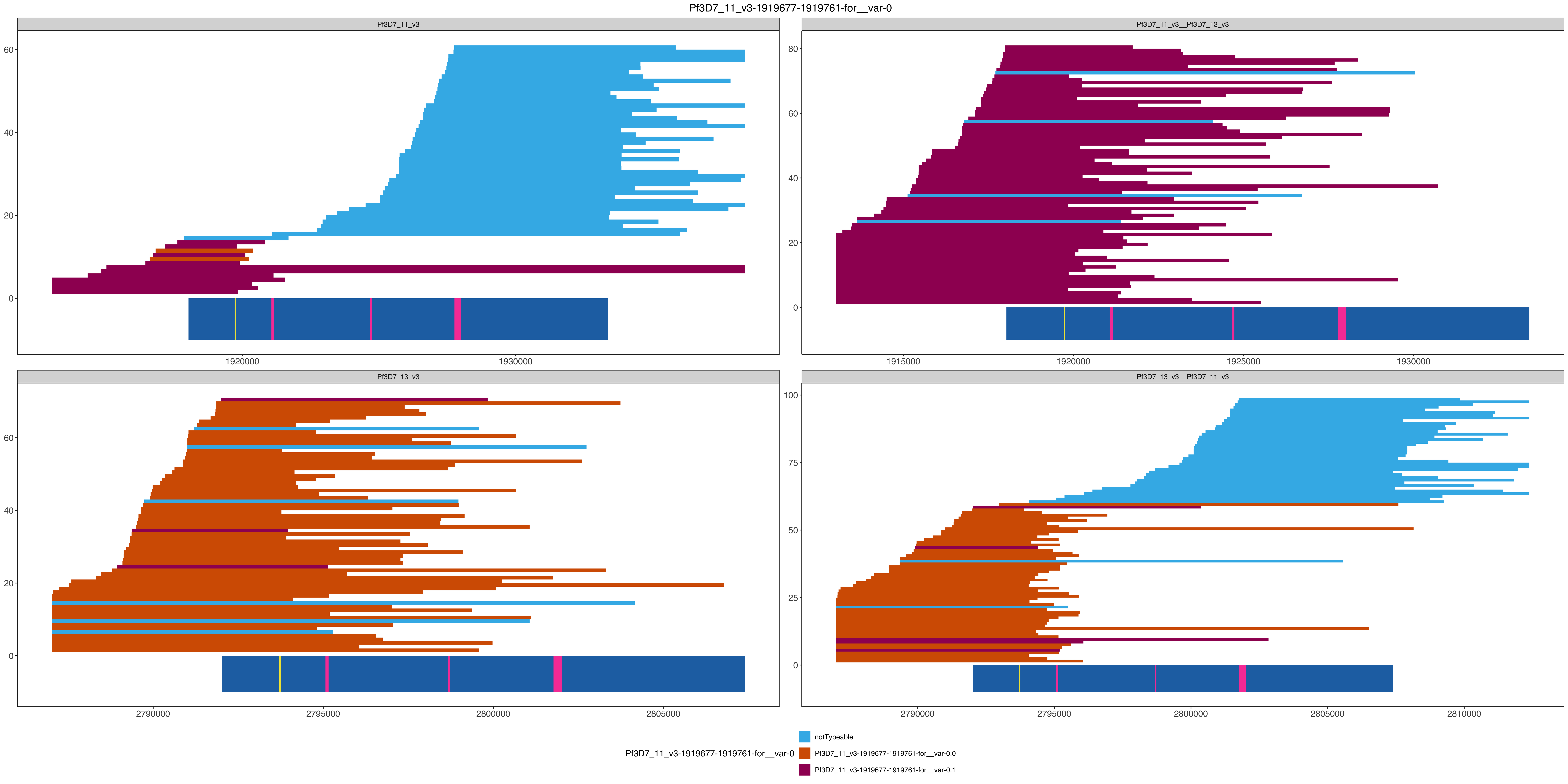
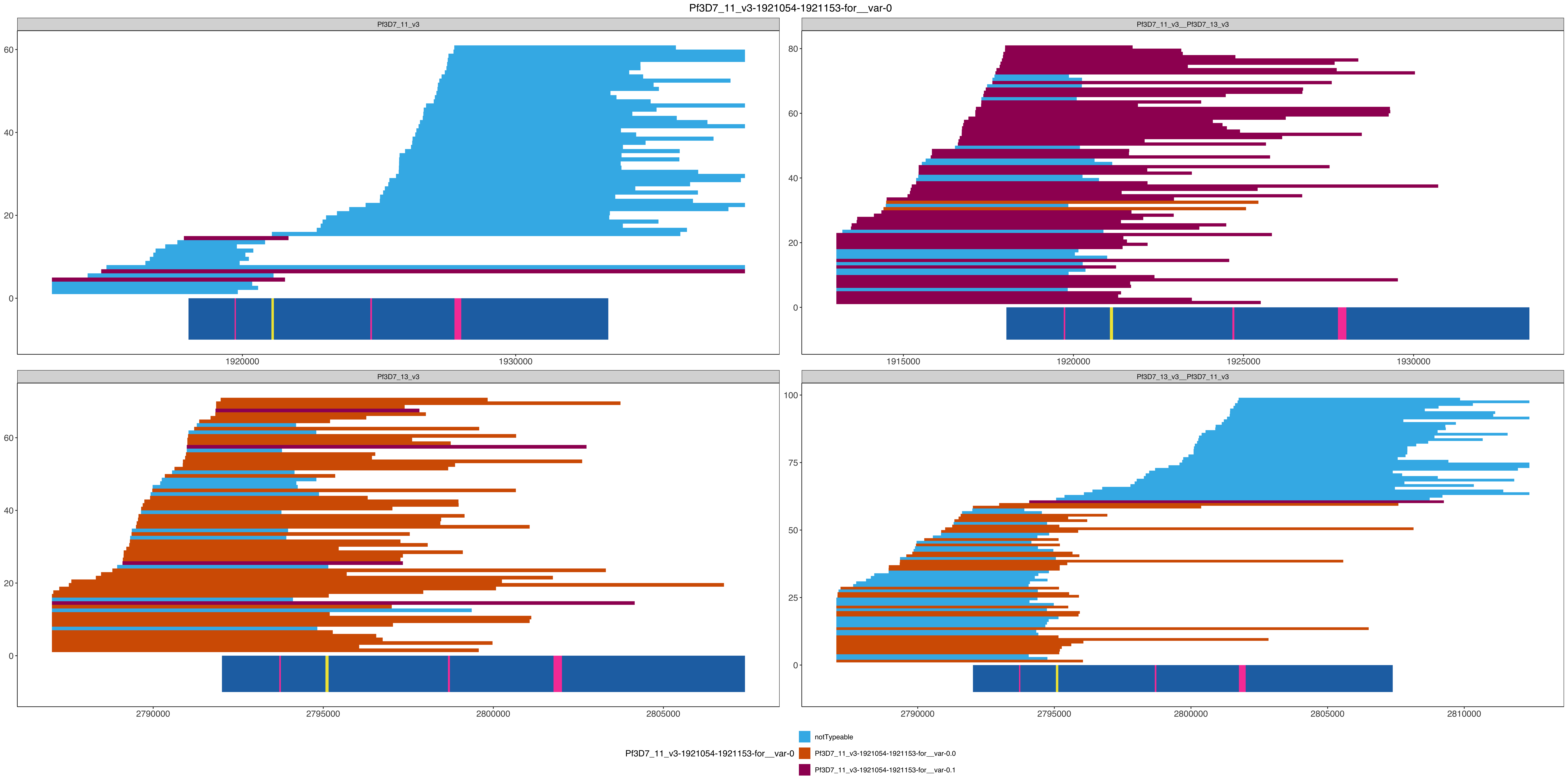
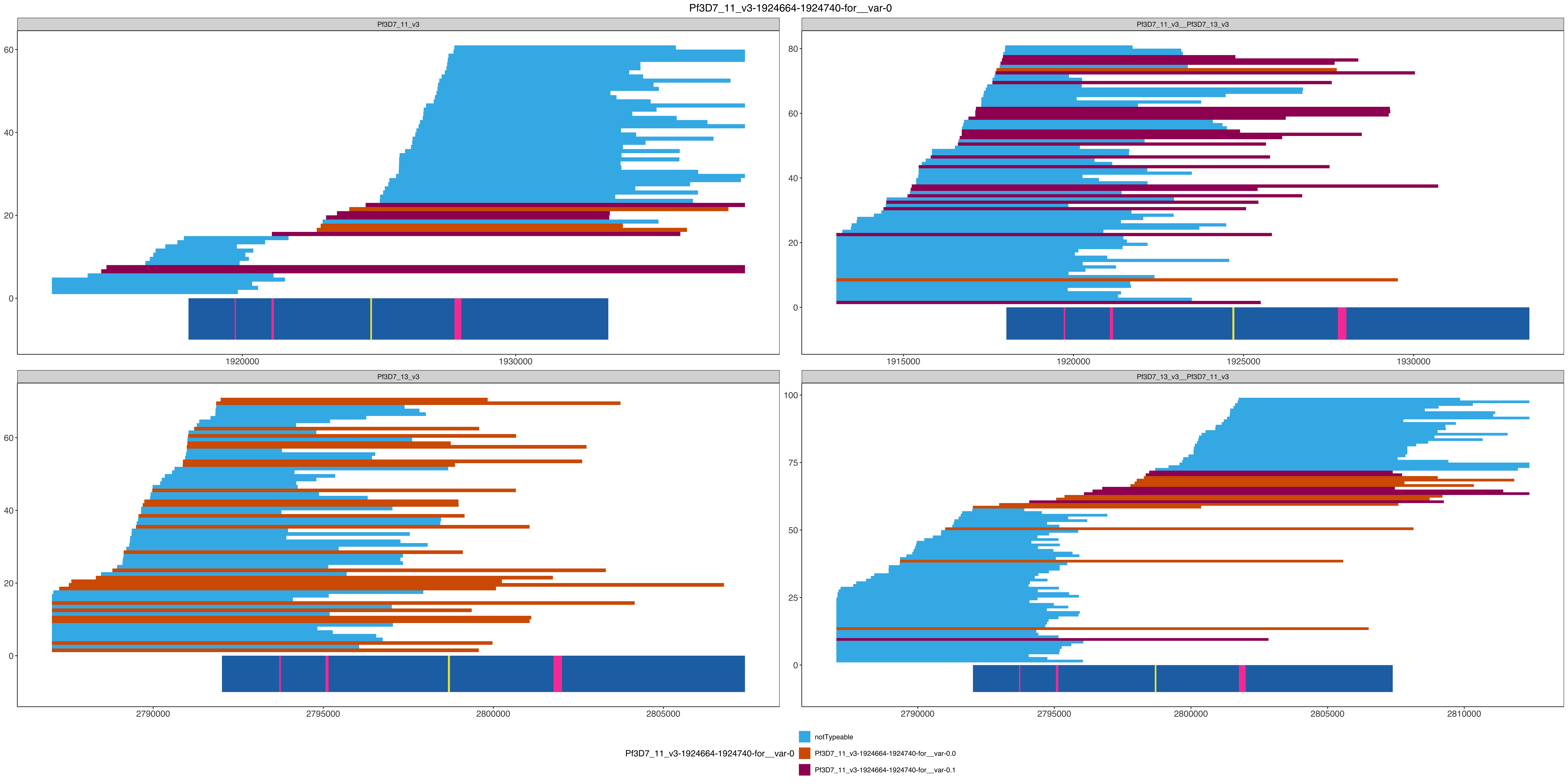
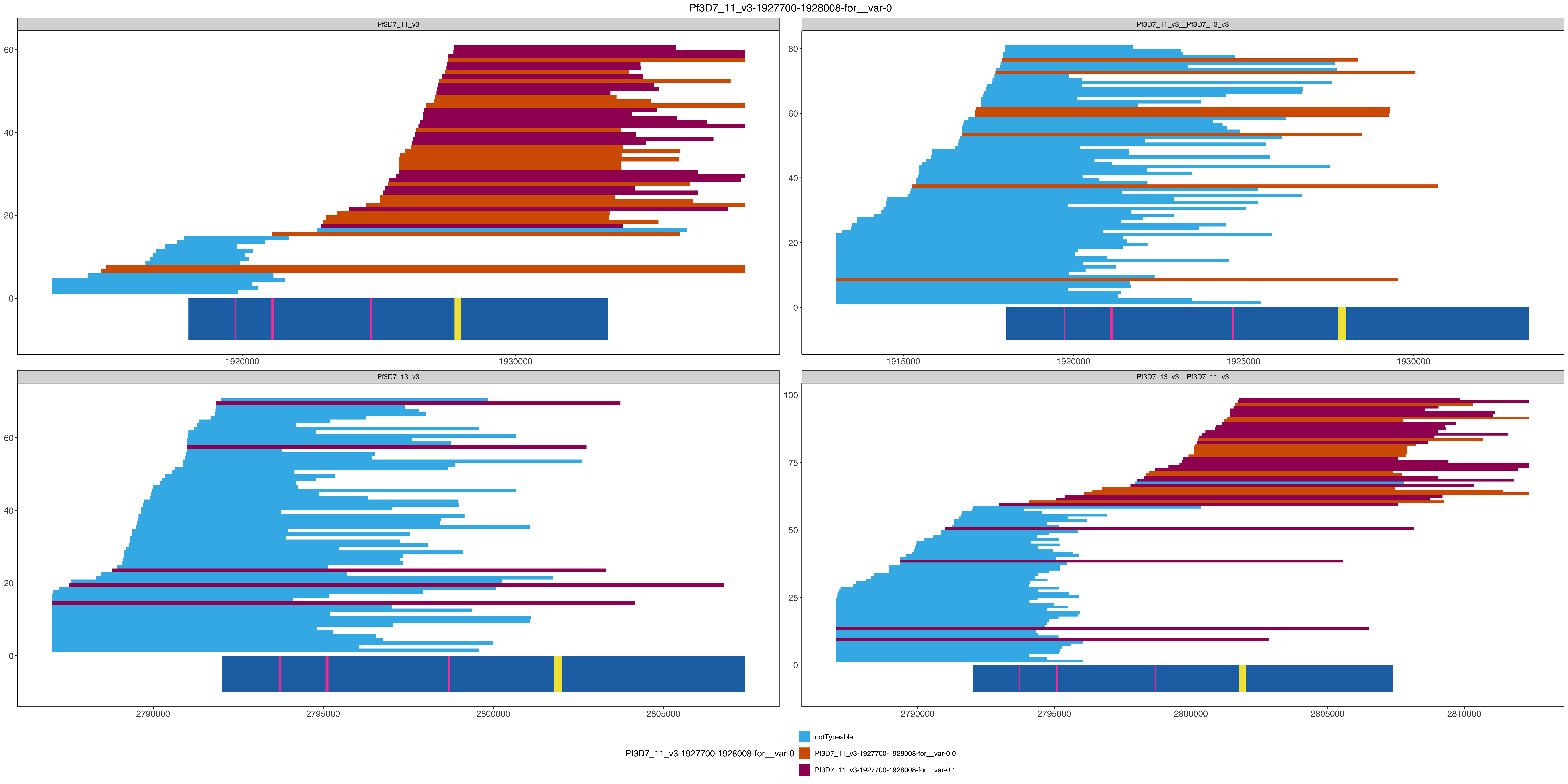
Typing the reads by the variants within each of the 4 regions, the current region typing the read by is colored pink in the shared region below. If a read didn’t cover the region or there was too much error to confidently type it then it’s colored as “untypeable”.
ggplot()+geom_rect(aes(xmin =start, xmax =end, ymin =rowid, ymax =rowid+1, fill =`Pf3D7_11_v3-1919677-1919761-for__var-0`), data =bothExtraction_filt_mod)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#2271B2", data =homologousRegionWithHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F748A5", data =multiRegion)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F0E442", data =multiRegion%>%filter(X4=="Pf3D7_11_v3-1919677-1919761-for__var-0"))+sofonias_theme+facet_wrap(~X1, scales ="free")+scale_fill_manual(values =c("#3DB7E9", "#D55E00", "#9F0162"), guide =guide_legend( ncol =1))+labs(title ="Pf3D7_11_v3-1919677-1919761-for__var-0")
Code
ggplot()+geom_rect(aes(xmin =start, xmax =end, ymin =rowid, ymax =rowid+1, fill =`Pf3D7_11_v3-1921054-1921153-for__var-0`), data =bothExtraction_filt_mod)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#2271B2", data =homologousRegionWithHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F748A5", data =multiRegion)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F0E442", data =multiRegion%>%filter(X4=="Pf3D7_11_v3-1921054-1921153-for__var-0"))+sofonias_theme+facet_wrap(~X1, scales ="free")+scale_fill_manual(values =c("#3DB7E9", "#D55E00", "#9F0162"), guide =guide_legend( ncol =1))+labs(title ="Pf3D7_11_v3-1921054-1921153-for__var-0")
Code
ggplot()+geom_rect(aes(xmin =start, xmax =end, ymin =rowid, ymax =rowid+1, fill =`Pf3D7_11_v3-1924664-1924740-for__var-0`), data =bothExtraction_filt_mod)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#2271B2", data =homologousRegionWithHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F748A5", data =multiRegion)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F0E442", data =multiRegion%>%filter(X4=="Pf3D7_11_v3-1924664-1924740-for__var-0"))+sofonias_theme+facet_wrap(~X1, scales ="free")+scale_fill_manual(values =c("#3DB7E9", "#D55E00", "#9F0162"), guide =guide_legend( ncol =1))+labs(title ="Pf3D7_11_v3-1924664-1924740-for__var-0")
Code
ggplot()+geom_rect(aes(xmin =start, xmax =end, ymin =rowid, ymax =rowid+1, fill =`Pf3D7_11_v3-1927700-1928008-for__var-0`), data =bothExtraction_filt_mod)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#2271B2", data =homologousRegionWithHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F748A5", data =multiRegion)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F0E442", data =multiRegion%>%filter(X4=="Pf3D7_11_v3-1927700-1928008-for__var-0"))+sofonias_theme+facet_wrap(~X1, scales ="free")+scale_fill_manual(values =c("#3DB7E9", "#D55E00", "#9F0162"), guide =guide_legend( ncol =1))+labs(title ="Pf3D7_11_v3-1927700-1928008-for__var-0")
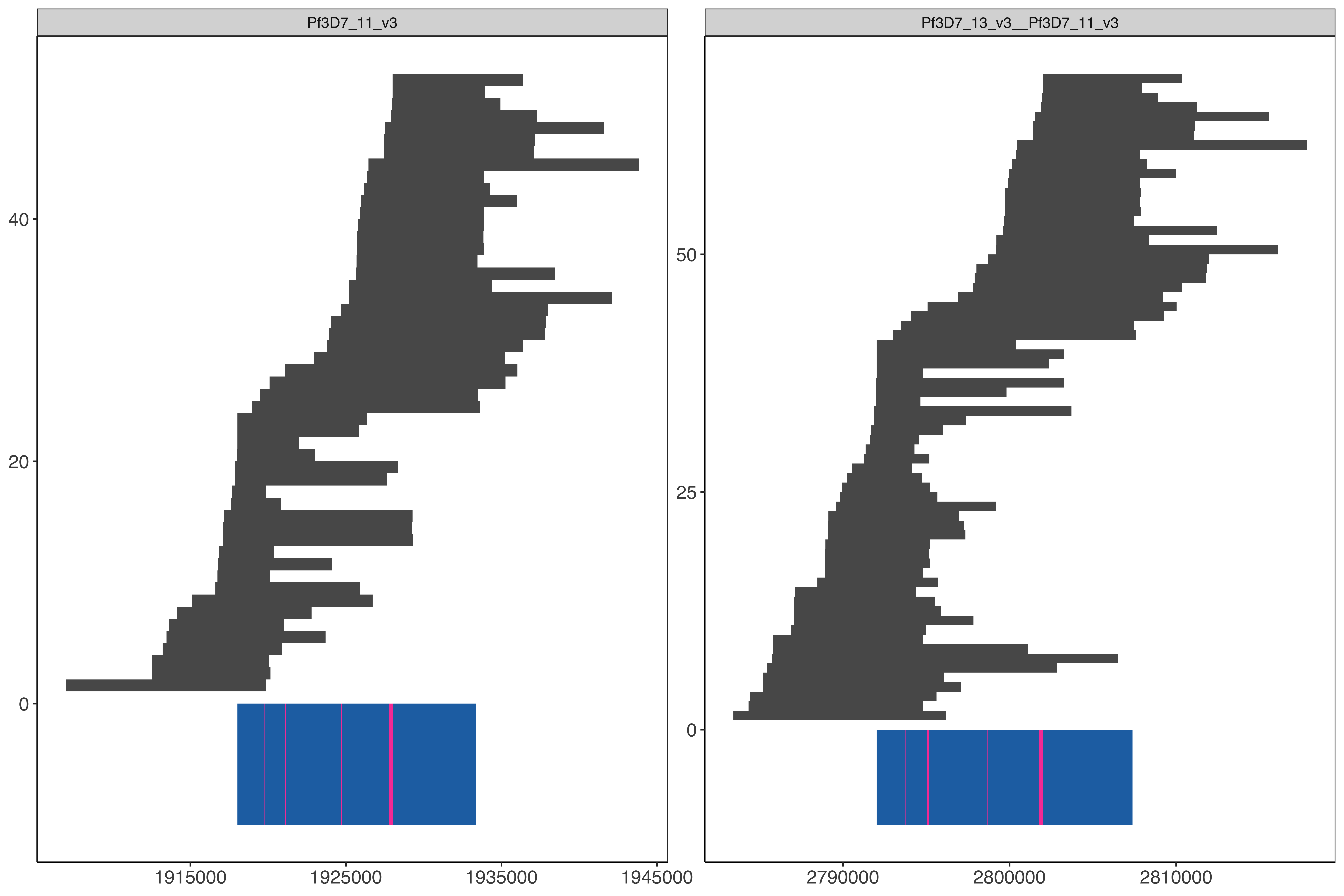
Processing on pure hybrid
The same exact work flow as above but except hybrid genome was modified to include only the expected regular chromosome 11 and the hybrid 13-11 (which is the expected hybrid genome for SD01). This way it is easier to deal with less multi mappers when so much genome was duplicated to map to.
Code
cd /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanoporemkdir purehybridAlnsMinimap2minimap2-x map-ont -t 40 -a /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_pure_11-13_13-11_hybrid.fasta rawFastq/PfSD01PermethION.fastq.gz |samtools sort --threads 40 -o purehybridAlnsMinimap2/PfSD01PermethION.sorted.bam samtools index purehybridAlnsMinimap2/PfSD01PermethION.sorted.bam cd /tank/data/plasmodium/falciparum/pfpubdata/malariagen_pacbio_assemblies mkdir purehybridAlnsMinimap2minimap2-x map-hifi -t 48 -a /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_pure_11-13_13-11_hybrid.fasta rawFastq/ERR1036246.fastq.gz |samtools sort --threads 48 -o purehybridAlnsMinimap2/PfSD01.sorted.bam &&samtools index purehybridAlnsMinimap2/PfSD01.sorted.bam
outBound=5000bothExtraction_filt_mod=bothExtraction_filt%>%group_by(`#chrom`)%>%arrange(`#chrom`, start, end)%>%mutate(rowid =row_number())%>%mutate(X1 =`#chrom`)%>%left_join(homologousRegionWithPureHybrid%>%rename(sharedStart =X2, sharedStop =X3)%>%select(X1, starts_with("shared")))%>%filter((start<sharedStart&end>sharedStart)|(start<sharedStop&end>sharedStop))%>%group_by(X1)%>%mutate(rowid =row_number())%>%group_by()%>%mutate(startDiff =sharedStart-start, endDiff =sharedStop-end)%>%mutate(start =ifelse(start<sharedStart-outBound, sharedStart-outBound, start), end =ifelse(end>sharedStop+outBound, sharedStop+outBound, end))illuminaExtraction_filt_mod=illuminaExtraction_filt%>%group_by(`#chrom`)%>%arrange(`#chrom`, start, end)%>%mutate(rowid =row_number())%>%mutate(X1 =`#chrom`)%>%left_join(homologousRegionWithPureHybrid%>%rename(sharedStart =X2, sharedStop =X3)%>%select(X1, starts_with("shared")))ggplot()+geom_rect(aes(xmin =start, xmax =end, ymin =rowid, ymax =rowid+1, fill =`Pf3D7_11_v3-1919677-1919761-for__var-0`), data =bothExtraction_filt_mod)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#2271B2", data =homologousRegionWithPureHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F748A5", data =multiRegionPureHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F0E442", data =multiRegionPureHybrid%>%filter(X4=="Pf3D7_11_v3-1919677-1919761-for__var-0"))+sofonias_theme+facet_wrap(~X1, scales ="free")+scale_fill_manual(values =c("#3DB7E9", "#D55E00", "#9F0162"), guide =guide_legend( ncol =1))+labs(title ="Pf3D7_11_v3-1919677-1919761-for__var-0")
Code
ggplot()+geom_rect(aes(xmin =start, xmax =end, ymin =rowid, ymax =rowid+1, fill =`Pf3D7_11_v3-1921054-1921153-for__var-0`), data =bothExtraction_filt_mod)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#2271B2", data =homologousRegionWithPureHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F748A5", data =multiRegionPureHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F0E442", data =multiRegionPureHybrid%>%filter(X4=="Pf3D7_11_v3-1921054-1921153-for__var-0"))+sofonias_theme+facet_wrap(~X1, scales ="free")+scale_fill_manual(values =c("#3DB7E9", "#D55E00", "#9F0162"), guide =guide_legend( ncol =1))+labs(title ="Pf3D7_11_v3-1921054-1921153-for__var-0")
Code
ggplot()+geom_rect(aes(xmin =start, xmax =end, ymin =rowid, ymax =rowid+1, fill =`Pf3D7_11_v3-1924664-1924740-for__var-0`), data =bothExtraction_filt_mod)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#2271B2", data =homologousRegionWithPureHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F748A5", data =multiRegionPureHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F0E442", data =multiRegionPureHybrid%>%filter(X4=="Pf3D7_11_v3-1924664-1924740-for__var-0"))+sofonias_theme+facet_wrap(~X1, scales ="free")+scale_fill_manual(values =c("#3DB7E9", "#D55E00", "#9F0162"), guide =guide_legend( ncol =1))+labs(title ="Pf3D7_11_v3-1924664-1924740-for__var-0")
Code
ggplot()+geom_rect(aes(xmin =start, xmax =end, ymin =rowid, ymax =rowid+1, fill =`Pf3D7_11_v3-1927700-1928008-for__var-0`), data =bothExtraction_filt_mod)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#2271B2", data =homologousRegionWithPureHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F748A5", data =multiRegionPureHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0), fill ="#F0E442", data =multiRegionPureHybrid%>%filter(X4=="Pf3D7_11_v3-1927700-1928008-for__var-0"))+sofonias_theme+facet_wrap(~X1, scales ="free")+scale_fill_manual(values =c("#3DB7E9", "#D55E00", "#9F0162"), guide =guide_legend( ncol =1))+labs(title ="Pf3D7_11_v3-1927700-1928008-for__var-0")
Linkage between variants
Looking at the linkage between variants by linking each variant within the long reads to each subsequent variant and then linkng the chromosome to the first variant. It appears that all the variants 0.1 are correlated to chromosome 11 and all variants 0.0 are correlated to chromosome 13
Code
# Load packagelibrary(networkD3)bothExtraction_filt_counts_gat=bothExtraction_filt_counts%>%gather(region, haplotype, 1:4)bothExtraction_filt_counts_gat_hapCounts=bothExtraction_filt_counts_gat%>%filter(haplotype!="notTypeable")%>%group_by(haplotype)%>%summarise(n =sum(n))%>%mutate(haplotypeCoded =row_number())bothExtraction_filt_counts_hapLinkCounts=tibble()# for(col1 in 1:3) {# for (col2 in (col1 + 1):4) {# bothExtraction_filt_counts_gat_subSelection = bothExtraction_filt_counts %>% # group_by() %>% # select(col1, col2, n)# colnames(bothExtraction_filt_counts_gat_subSelection)[1:2] = c("hap1", "hap2")# bothExtraction_filt_counts_gat_subSelection_filt = bothExtraction_filt_counts_gat_subSelection %>% # filter(hap1 != "notTypeable", # hap2 != "notTypeable") %>% # group_by(hap1, hap2) %>% # summarise(n = sum(n))# bothExtraction_filt_counts_hapLinkCounts = bind_rows(# bothExtraction_filt_counts_hapLinkCounts, # bothExtraction_filt_counts_gat_subSelection_filt# )# }# }for(col1in1:3){col2=col1+1bothExtraction_filt_counts_gat_subSelection=bothExtraction_filt_counts%>%group_by()%>%select(col1, col2, n)colnames(bothExtraction_filt_counts_gat_subSelection)[1:2]=c("hap1", "hap2")bothExtraction_filt_counts_gat_subSelection_filt=bothExtraction_filt_counts_gat_subSelection%>%filter(hap1!="notTypeable", hap2!="notTypeable")%>%group_by(hap1, hap2)%>%summarise(n =sum(n))bothExtraction_filt_counts_hapLinkCounts=bind_rows(bothExtraction_filt_counts_hapLinkCounts, bothExtraction_filt_counts_gat_subSelection_filt)}bothExtraction_filt_chromosomeLinkageInfo=bothExtraction_filt%>%select(`#chrom`, `Pf3D7_11_v3-1919677-1919761-for__var-0`)%>%filter(`Pf3D7_11_v3-1919677-1919761-for__var-0`!="notTypeable")%>%group_by(`#chrom`, `Pf3D7_11_v3-1919677-1919761-for__var-0`)%>%count()bothExtraction_filt_chromosomeLinkageInfo_addToHapCounts=bothExtraction_filt_chromosomeLinkageInfo%>%rename(haplotype =`#chrom`)%>%group_by(haplotype)%>%summarise(n =sum(n))bothExtraction_filt_counts_gat_hapCounts=bothExtraction_filt_counts_gat_hapCounts%>%bind_rows(bothExtraction_filt_chromosomeLinkageInfo_addToHapCounts)#zero based position source/target nodes bothExtraction_filt_counts_hapLinkCounts=bothExtraction_filt_counts_hapLinkCounts%>%mutate(source =match(hap1, bothExtraction_filt_counts_gat_hapCounts$haplotype)-1)%>%mutate(target =match(hap2, bothExtraction_filt_counts_gat_hapCounts$haplotype)-1)bothExtraction_filt_chromosomeLinkageInfo=bothExtraction_filt_chromosomeLinkageInfo%>%rename(hap1 =`#chrom`, hap2 =`Pf3D7_11_v3-1919677-1919761-for__var-0`)%>%mutate(source =match(hap1, bothExtraction_filt_counts_gat_hapCounts$haplotype)-1)%>%mutate(target =match(hap2, bothExtraction_filt_counts_gat_hapCounts$haplotype)-1)bothExtraction_filt_counts_hapLinkCounts=bothExtraction_filt_counts_hapLinkCounts%>%bind_rows(bothExtraction_filt_chromosomeLinkageInfo)# adding in chromosome linkage to front bothExtraction_filt_counts_hapLinkCounts=as.data.frame(bothExtraction_filt_counts_hapLinkCounts)bothExtraction_filt_counts_gat_hapCounts=as.data.frame(bothExtraction_filt_counts_gat_hapCounts)# Thus we can plot itp<-sankeyNetwork( Links =bothExtraction_filt_counts_hapLinkCounts, Nodes =bothExtraction_filt_counts_gat_hapCounts, Source ="source", Target ="target", Value ="n", NodeID ="haplotype", units ="reads", fontSize =12, nodeWidth =30)
There is some cross linkage between the variants but this is low level and could either represent typing error or even possibly low level of continued recombination between these regions.
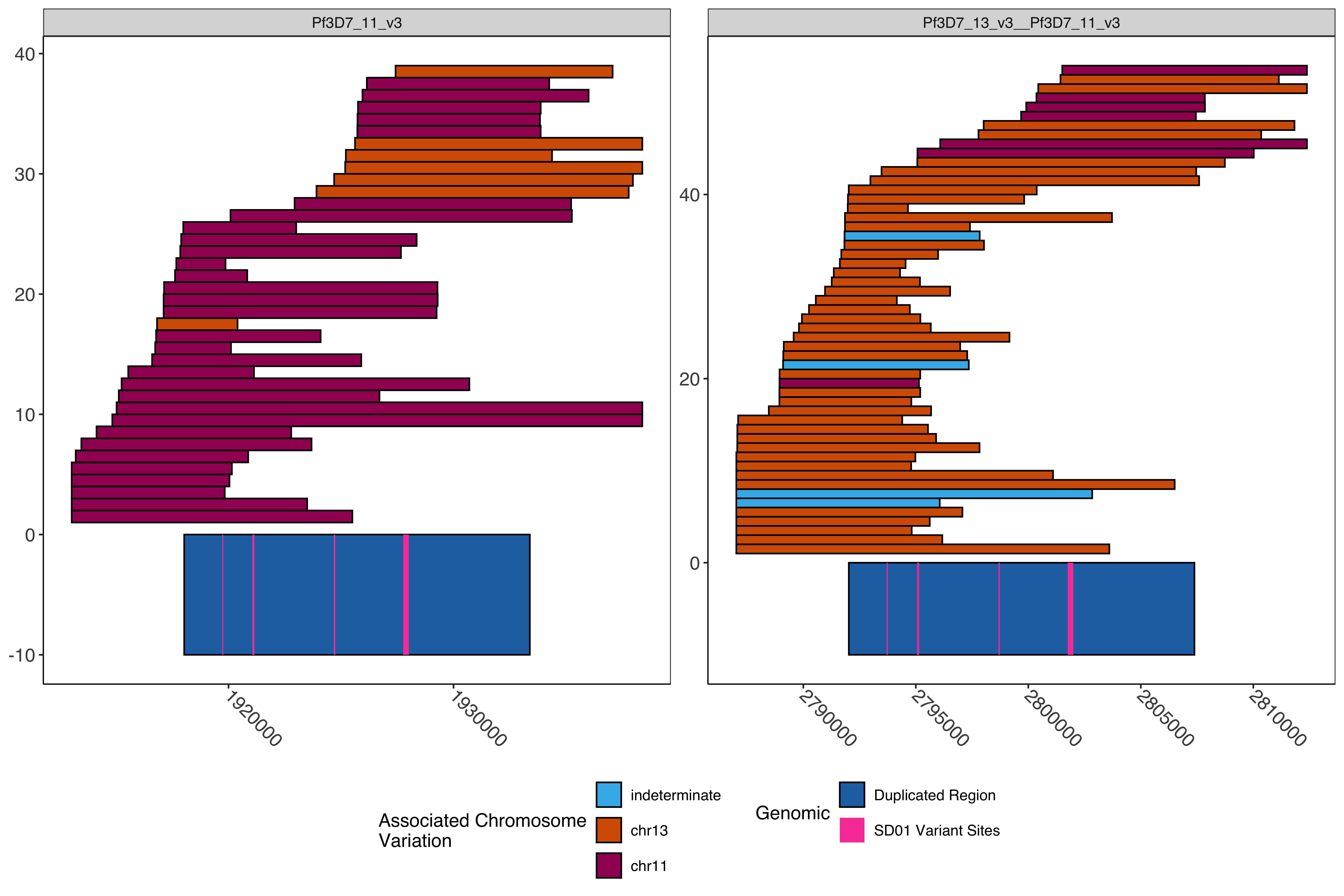
Colored by the chromosome associated variation (chr11 vs chr13). If a read has multiple loci and there is a tie between the different chromosomes then it is colored indeterminate.
Code
chrVarPlot=ggplot()+geom_rect(aes(xmin =start, xmax =end, ymin =rowid, ymax =rowid+1, fill =typed), color ="black", data =bothExtraction_filt_mod_typed%>%mutate(typed =factor(typed, levels =c("indeterminate", "chr13", "chr11"))))+scale_fill_manual(values =c("#3DB7E9", "#D55E00", "#9F0162"), name="Associated Chromosome\nVariation", guide =guide_legend( ncol =1))+ggnewscale::new_scale_fill()+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0, fill ="Duplicated Region"), color ="black",#fill = "#2271B2", data =homologousRegionWithPureHybrid)+geom_rect(aes(xmin =X2, xmax =X3, ymin =-10, ymax =0, fill ="SD01 Variant Sites"),#color = "black",#fill = "#F748A5", data =multiRegionPureHybrid)+scale_fill_manual("Genomic", values =c("Duplicated Region"="#2271B2", "SD01 Variant Sites"="#F748A5"))+guides(fill =guide_legend(ncol =1))+sofonias_theme+theme(axis.text.x =element_text(size=12, angle =-45, vjust =0.5, hjust =0))+facet_wrap(~X1, scales ="free")print(chrVarPlot)