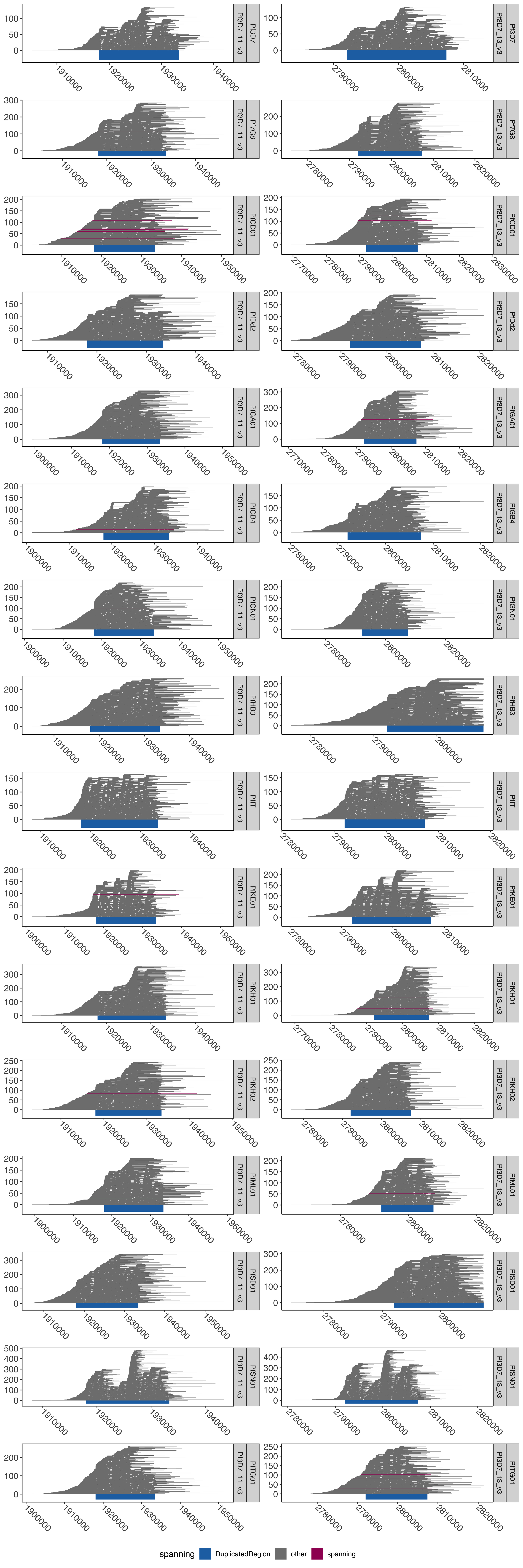
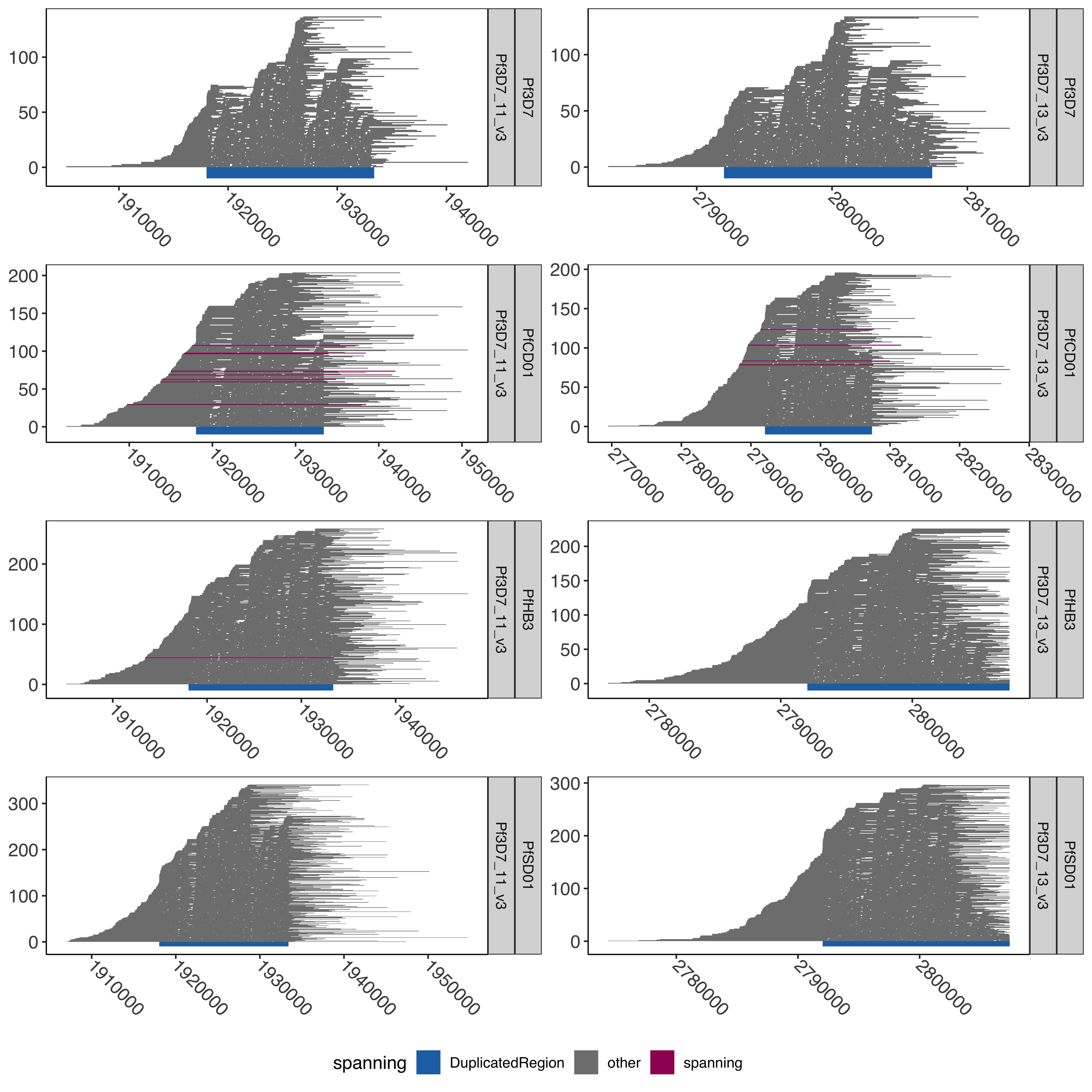
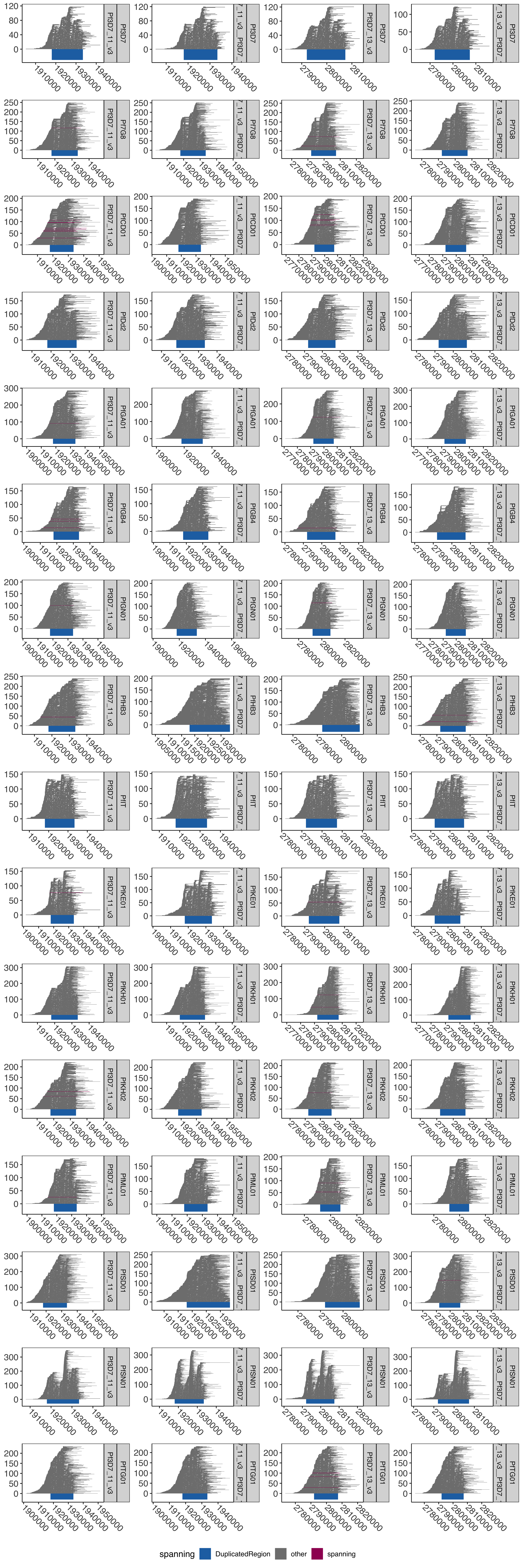
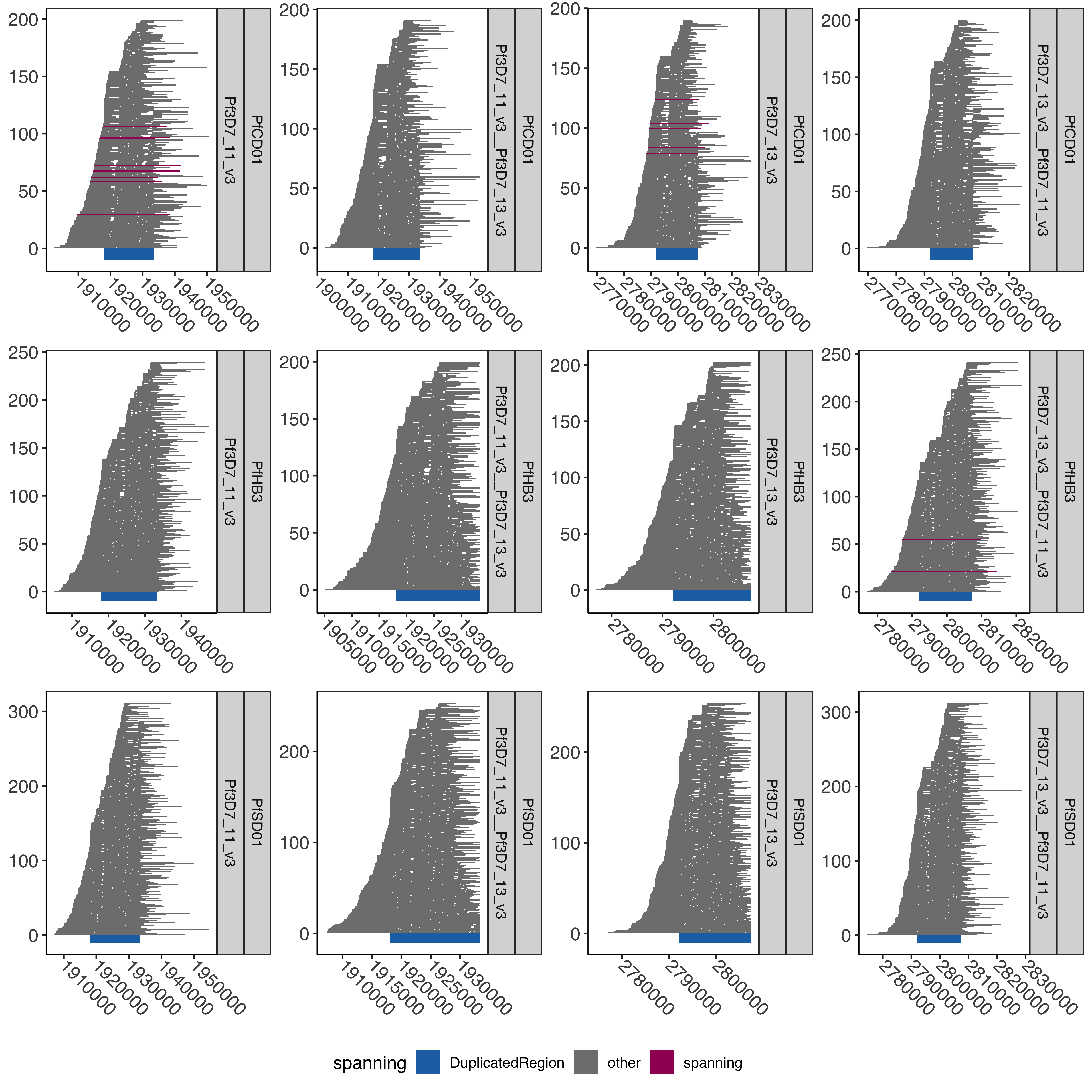
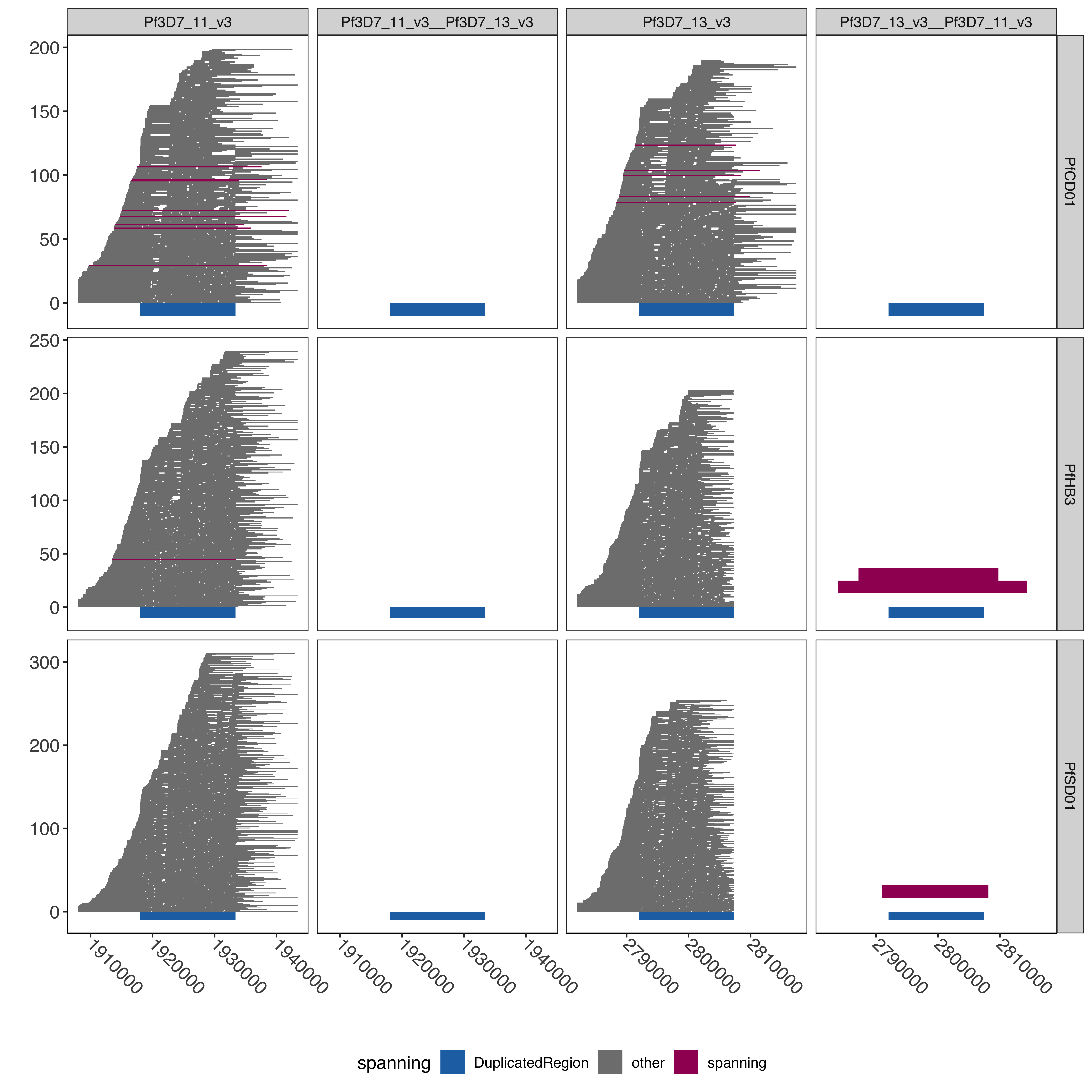
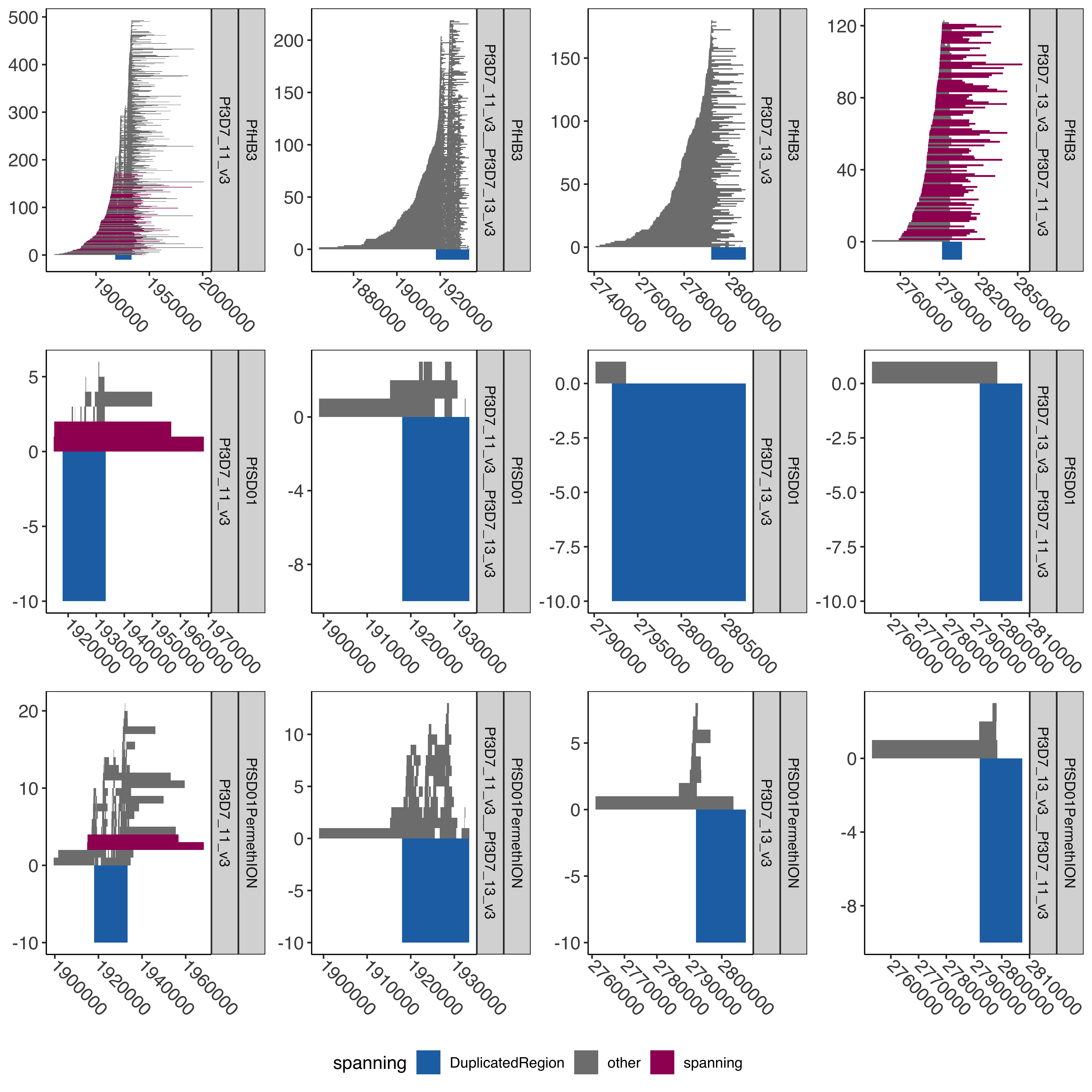
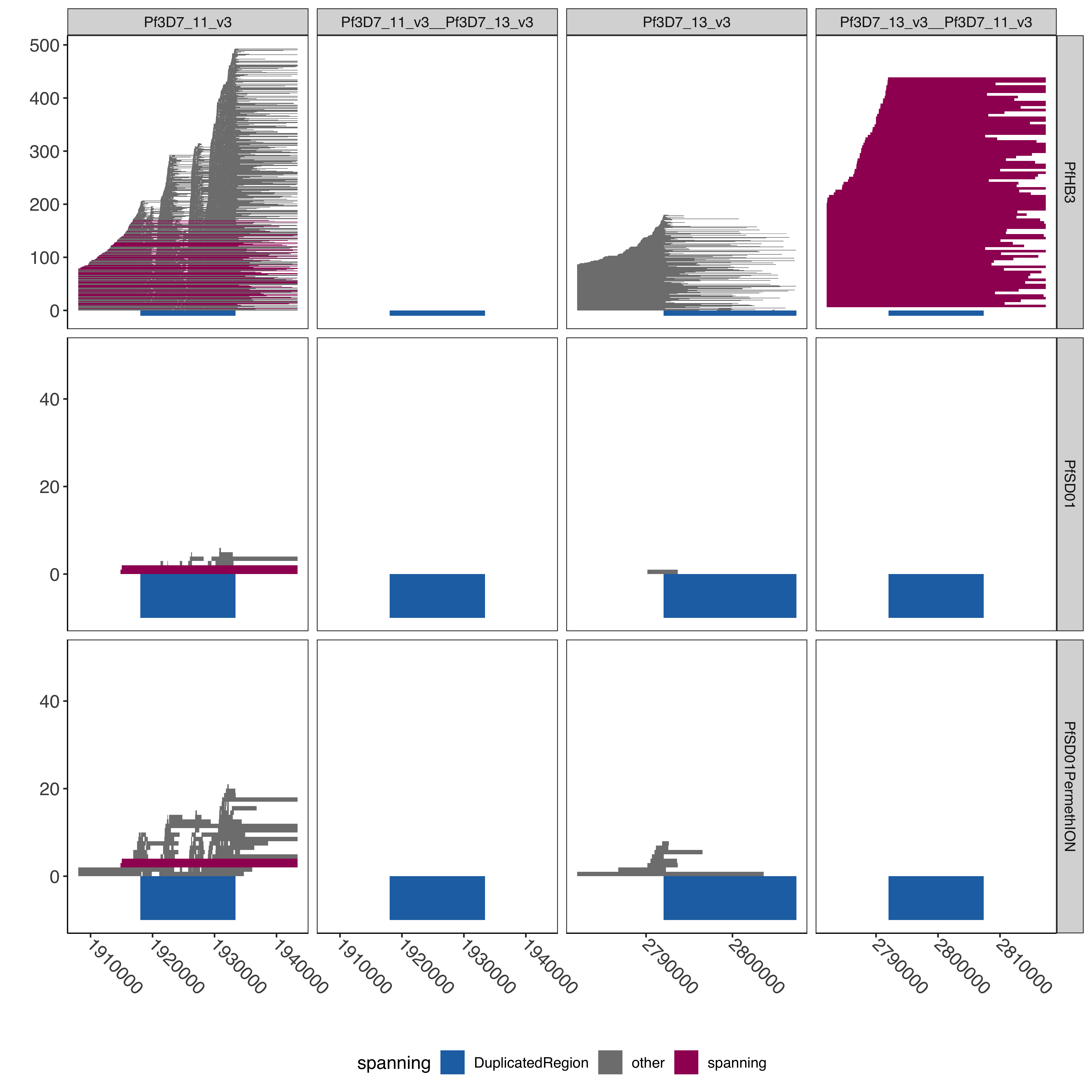
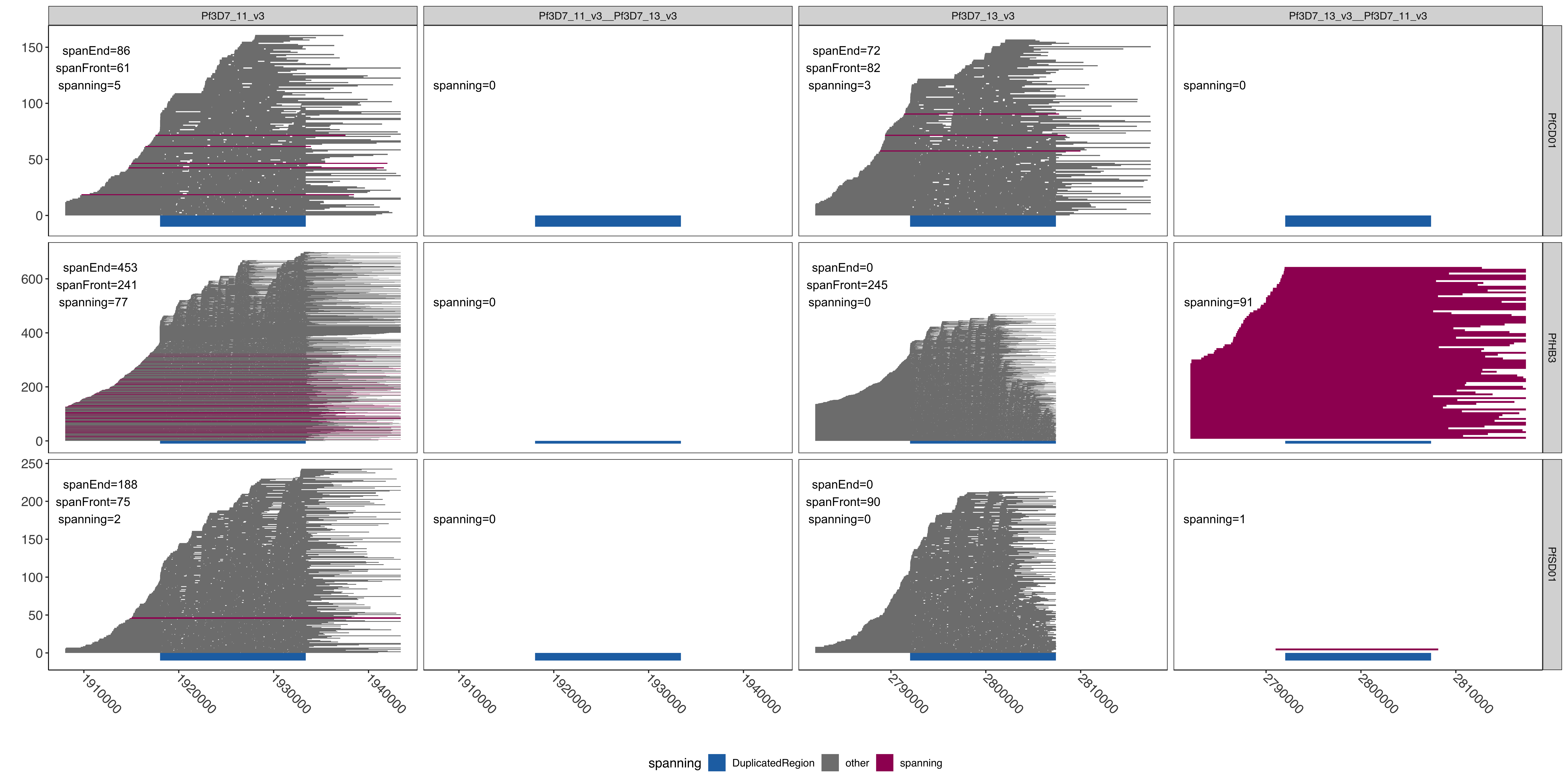
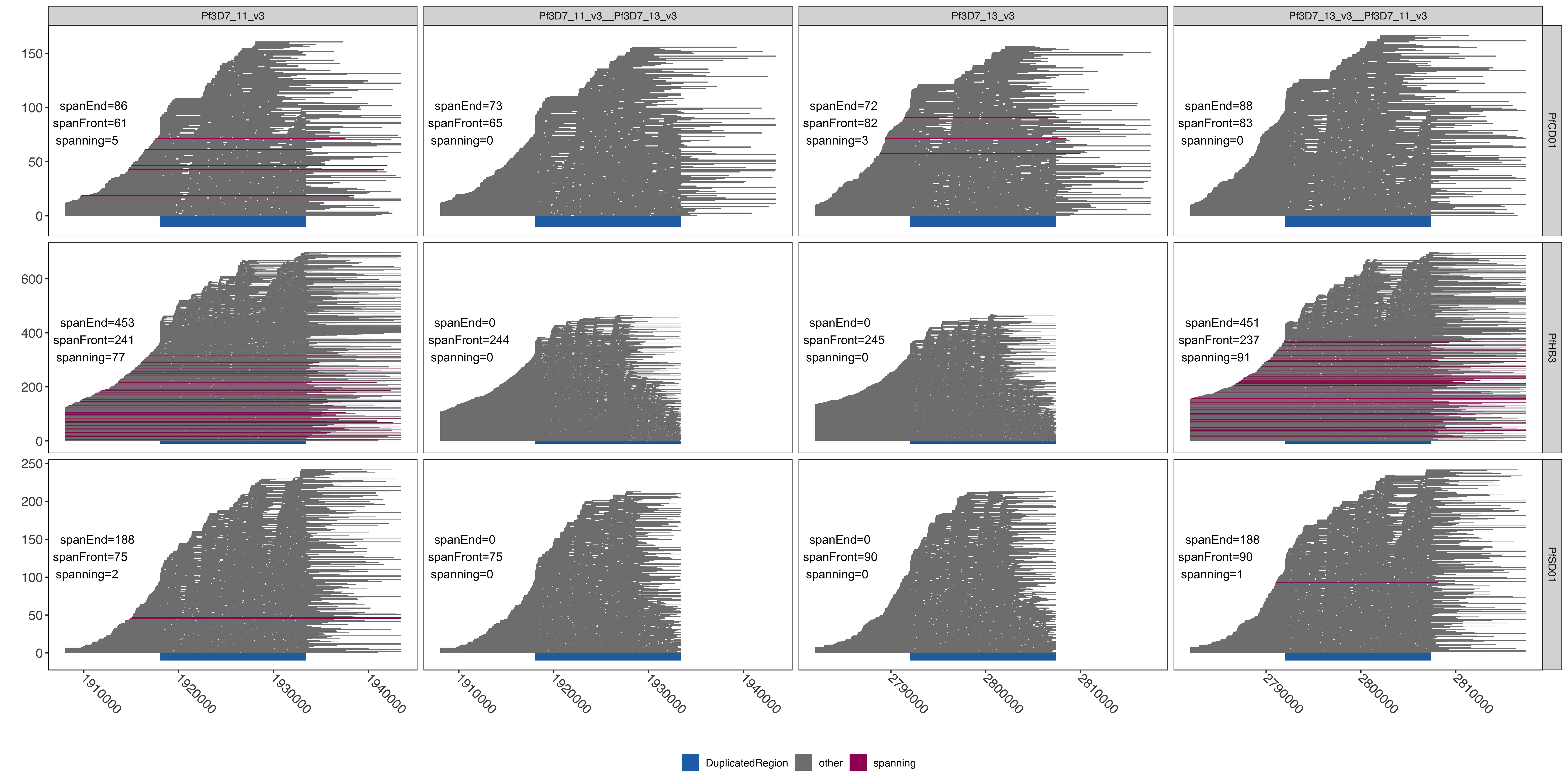
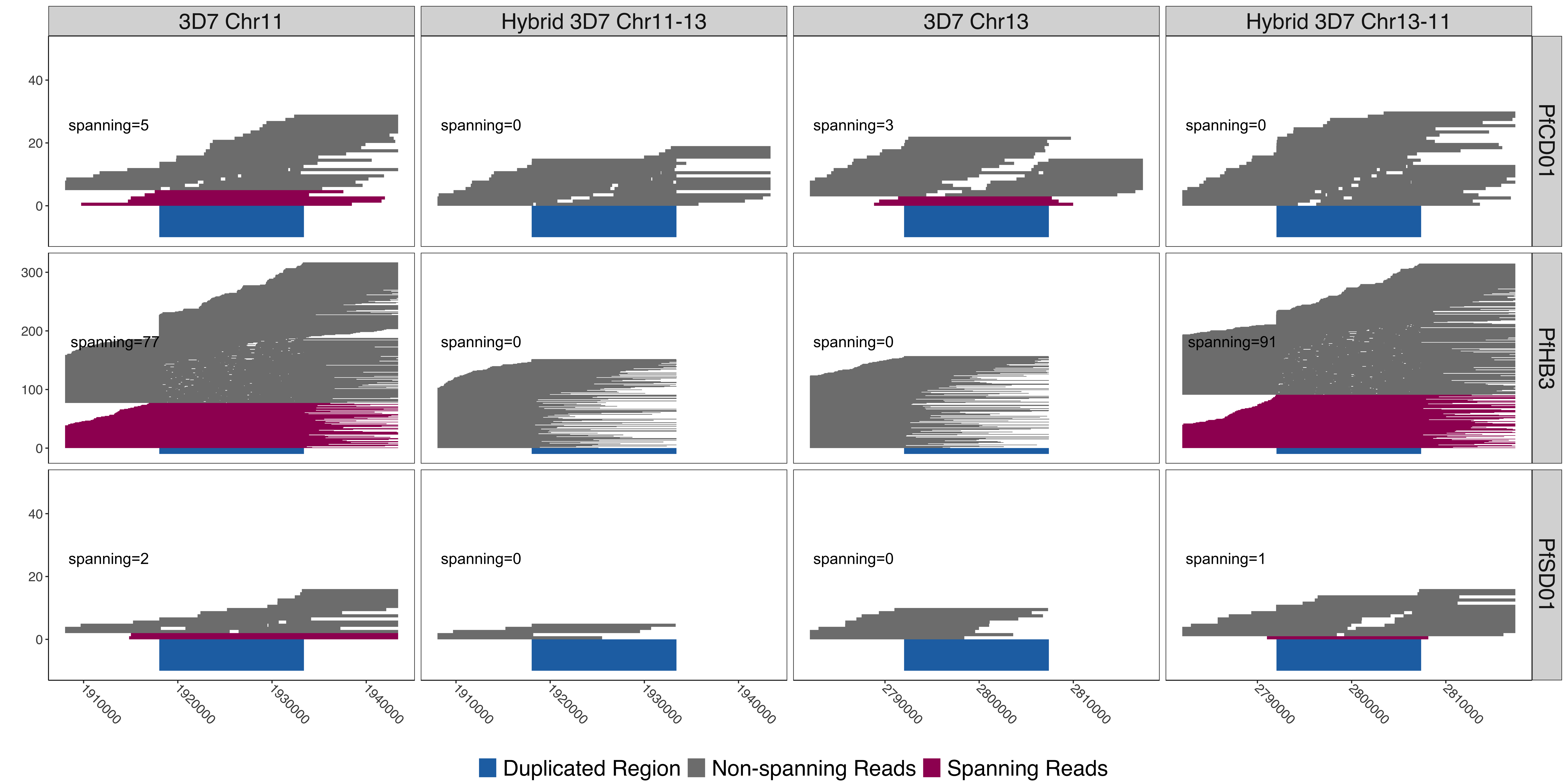
--- title: "Processing Spanning Reads" code-fold: true --- ```{r setup, echo=FALSE, message=FALSE} source("../../common.R") ``` # Processing Spanning reads across shared region in control samples ## Creating hybrid chr13/chr11 genome ```{bash, eval= F} elucidator extractByName --names "Pf3D7_11_v3 | organism=Plasmodium_falciparum_3D7 | version=2015-06-18 | length=2038340 | SO=chromosome" --fasta /tank/data/plasmodium/genomes/pf/genomes/Pf3D7.fasta --out Pf3D7_11_v3.fasta --overWrite elucidator extractByName --names "Pf3D7_13_v3 | organism=Plasmodium_falciparum_3D7 | version=2015-06-18 | length=2925236 | SO=chromosome" --fasta /tank/data/plasmodium/genomes/pf/genomes/Pf3D7.fasta --out Pf3D7_13_v3.fasta --overWrite elucidator trimToLen --fasta Pf3D7_11_v3.fasta --length 1918029 --overWrite --out Pf3D7_11_v3_front.fasta elucidator trimToLen --fasta Pf3D7_13_v3.fasta --length 2792022 --overWrite --out Pf3D7_13_v3_front.fasta elucidator trimFront --fasta Pf3D7_11_v3.fasta --forwardBases 1918029 --overWrite --out Pf3D7_11_v3_end.fasta elucidator trimFront --fasta Pf3D7_13_v3.fasta --forwardBases 2792022 --overWrite --out Pf3D7_13_v3_end.fasta elucidator appendReads --fasta Pf3D7_11_v3_front.fasta --seq Pf3D7_13_v3_end.fasta --overWrite --out Pf3D7_11_v3__Pf3D7_13_v3.fasta elucidator appendReads --fasta Pf3D7_13_v3_front.fasta --seq Pf3D7_11_v3_end.fasta --overWrite --out Pf3D7_13_v3__Pf3D7_11_v3.fasta sed 's/ .*/__Pf3D7_11_v3/g' Pf3D7_13_v3__Pf3D7_11_v3.fasta > renamed_Pf3D7_13_v3__Pf3D7_11_v3.fasta sed 's/ .*/__Pf3D7_13_v3/g' Pf3D7_11_v3__Pf3D7_13_v3.fasta > renamed_Pf3D7_11_v3__Pf3D7_13_v3.fasta elucidator sortReads --fasta combined_Pf3D7.fasta --sortBy name --overWrite --out Pf3D7_plus_11-13_13-11_hybrid.fasta ``` ```{r} = readr:: read_tsv ("../../../sharedBetween11_and_13/investigatingChrom11Chrom13/shared_11_13_region.bed" , col_names = T)= shared_11_13_region %>% mutate (` #chrom ` = gsub ("^Pf3D7_11_v3$" , "Pf3D7_11_v3__Pf3D7_13_v3" , ` #chrom ` ))%>% mutate (` #chrom ` = gsub ("^Pf3D7_13_v3$" , "Pf3D7_13_v3__Pf3D7_11_v3" , ` #chrom ` )) %>% mutate (end = ifelse (` #chrom ` == "Pf3D7_11_v3__Pf3D7_13_v3" , start + 15375 , end)) %>% mutate (end = ifelse (` #chrom ` == "Pf3D7_13_v3__Pf3D7_11_v3" , start + 15361 , end)) %>% mutate (len = end - start)write_tsv (shared_11_13_region_hybrid, "shared_11_13_region_onHybrid.bed" )``` ## Process Control files [ @Otto2018-bb ] . These samples have suspected transposition based on coverage data(no chr13 sub-telomere coverage and double chr11 sub-telomere coverage). There is also data from 13 other lab/clinical isolates. ## PfSD01 and PfHB3 were nanopored ### BWA mem #### Regular Pf3D7 genome file ```{bash, eval = F} CMD:if [ ! -f bams/{SAMPLE}.sorted.bam.bai ]; then bwa mem -v 1 -x ont2d -t {NUMTHREADS} /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta rawFastq/{SAMPLE}.fastq.gz| samtools sort --threads {NUMTHREADS} -o bams/{SAMPLE}.sorted.bam && samtools index bams/{SAMPLE}.sorted.bam; else echo {SAMPLE} done; fi; {SAMPLE}:samples.txt {NUMTHREADS}:4 ``` #### Chr11-13 hybrid Pf3D7 genome file ```{bash, eval = F} CMD:if [ ! -f hybridAlns/{SAMPLE}.sorted.bam.bai ]; then bwa mem -v 1 -x ont2d -t {NUMTHREADS} /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta rawFastq/{SAMPLE}.fastq.gz| samtools sort --threads {NUMTHREADS} -o hybridAlns/{SAMPLE}.sorted.bam && samtools index hybridAlns/{SAMPLE}.sorted.bam; else echo {SAMPLE} done; fi; {SAMPLE}:samples.txt {NUMTHREADS}:4 ``` ### minimap2 #### Regular Pf3D7 genome file ```{bash, eval = F} CMD:if [ ! -f bamsMinimap2/{SAMPLE}.sorted.bam.bai ]; then minimap2 -x map-ont -t {NUMTHREADS} -a /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta rawFastq/{SAMPLE}.fastq.gz | samtools sort --threads {NUMTHREADS} -o bamsMinimap2/{SAMPLE}.sorted.bam && samtools index bamsMinimap2/{SAMPLE}.sorted.bam; else echo {SAMPLE} done; fi; {SAMPLE}:samples.txt {NUMTHREADS}:4 ``` ```{bash, eval = F} PREP:mkdir -p bamsMinimap2AgainstPfSD01 CMD:if [ ! -f bamsMinimap2AgainstPfSD01/{SAMPLE}.sorted.bam.bai ]; then minimap2 -x map-ont -t {NUMTHREADS} -a /tank/data/genomes/plasmodium/genomes/pf_others/genomes/PfSD01.fasta rawFastq/{SAMPLE}.fastq.gz | samtools sort --threads {NUMTHREADS} -o bamsMinimap2AgainstPfSD01/{SAMPLE}.sorted.bam && samtools index bamsMinimap2AgainstPfSD01/{SAMPLE}.sorted.bam; else echo {SAMPLE} done; fi; {SAMPLE}:PfSD01,PfSD01PermethION {NUMTHREADS}:4 ``` ```{bash, eval = F} bamtools merge -in PfSD01PermethION.sorted.bam -in PfSD01.sorted.bam -out PfSD01Combined.sorted.bam && samtools index PfSD01Combined.sorted.bam ``` #### Chr11-13 hybrid Pf3D7 genome file ```{bash, eval = F} CMD:if [ ! -f hybridAlnsMinimap2/{SAMPLE}.sorted.bam.bai ]; then minimap2 -x map-ont -t {NUMTHREADS} -a /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta rawFastq/{SAMPLE}.fastq.gz| samtools sort --threads {NUMTHREADS} -o hybridAlnsMinimap2/{SAMPLE}.sorted.bam && samtools index hybridAlnsMinimap2/{SAMPLE}.sorted.bam; else echo {SAMPLE} done; fi; {SAMPLE}:samples.txt {NUMTHREADS}:4 ``` ### Intersecting with shared region ```{bash, eval = F} rm -f bamHybridMinimap2IntersectCmds.txt && for x in hybridAlnsMinimap2/Pf*bai; do if [ ! -f $(basename ${x%%.sorted.bam.bai})_nanopore_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region.bed ]; then echo "elucidator bamToBed --bam ${x%%.bai} | elucidator getOverlappingBedRegions --bed STDIN --intersectWithBed combined_shared_11_13_region.bed --out $(basename ${x%%.sorted.bam.bai})_nanopore_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region.bed" >> bamHybridMinimap2IntersectCmds.txt; fi ; done; ``` ## Previous Pacbio reads ```{bash, eval = F} elucidator printCol --file /tank/data/genomes/plasmodium/genomes/pf/info/rawDataSRRAccession.tab.txt --delim tab --header --columnName Pacbio > pacbio_accs.txt getSraRunsFromSampleAcc.py --identifiers pacbio_accs.txt --outStub pacbio mkdir sra rawFastq bams hybridAlns hybridAlnsMinimap2 bamsMinimap2 ``` ### Download pacbio reads ```{bash, eval = F} cd sra nohup downloadSraFromTable.py --sraUrlFnp ../pacbio_urls.tab.txt --urlColumn amazon_url --ncpus 5 & ls *.sra | sed 's/.sra//g' > ../samples.txt ``` ### Extract fastq reads ```{bash, eval = F} rm -fr runFastqDumpRawCmds.txt && for x in sra/*.sra; do echo fastq-dump ${x} --outdir rawFastq/ >> runFastqDumpRawCmds.txt ; done; nohup elucidator runMultipleCommands --cmdFile runFastqDumpRawCmds.txt --numThreads 1 --raw & for x in *.fastq; do echo ${x} && pigz -p 40 ${x}; done; ``` ```{r} = readr:: read_tsv ("/tank/data/genomes/plasmodium/genomes/pf/info/rawDataSRRAccession.tab.txt" , show_col_types = F)= readr:: read_tsv ("pacbio_info.tab.txt" , show_col_types = F)= sampleToAcc %>% left_join (accInfo %>% select (Run, Sample) %>% rename (Pacbio = Sample))``` ### BWA mem #### Regular Pf3D7 genome file ```{bash, eval = F} CMD:if [ ! -f bams/{SAMPLE}.sorted.bam.bai ]; then bwa mem -v 1 -x pacbio -t {NUMTHREADS} /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta rawFastq/{SAMPLE}.fastq.gz| samtools sort -o bams/{SAMPLE}.sorted.bam && samtools index bams/{SAMPLE}.sorted.bam; else echo {SAMPLE} done; fi; {SAMPLE}:samples.txt {NUMTHREADS}:4 ``` #### Chr11-13 hybrid Pf3D7 genome file ```{bash, eval = F} CMD:if [ ! -f hybridAlns/{SAMPLE}.sorted.bam.bai ]; then bwa mem -v 1 -x pacbio -t {NUMTHREADS} /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta rawFastq/{SAMPLE}.fastq.gz| samtools sort -o hybridAlns/{SAMPLE}.sorted.bam && samtools index hybridAlns/{SAMPLE}.sorted.bam; else echo {SAMPLE} done; fi; {SAMPLE}:samples.txt {NUMTHREADS}:4 ``` ### minimap2 #### Regular Pf3D7 genome file ```{bash, eval = F} CMD:if [ ! -f bamsMinimap2/{SAMPLE}.sorted.bam.bai ]; then minimap2 -x map-hifi -t {NUMTHREADS} -a /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta rawFastq/{SAMPLE}.fastq.gz | samtools sort -o bamsMinimap2/{SAMPLE}.sorted.bam && samtools index bamsMinimap2/{SAMPLE}.sorted.bam; else echo {SAMPLE} done; fi; {SAMPLE}:samples.txt {NUMTHREADS}:4 ``` #### Chr11-13 hybrid Pf3D7 genome file ```{bash, eval = F} CMD:if [ ! -f hybridAlnsMinimap2/{SAMPLE}.sorted.bam.bai ]; then minimap2 -x map-hifi -t {NUMTHREADS} -a /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta rawFastq/{SAMPLE}.fastq.gz| samtools sort -o hybridAlnsMinimap2/{SAMPLE}.sorted.bam && samtools index hybridAlnsMinimap2/{SAMPLE}.sorted.bam; else echo {SAMPLE} done; fi; {SAMPLE}:samples.txt {NUMTHREADS}:4 ``` ### merge ```{r} = readr:: read_tsv ("rawDataSRRAccession.tab.txt" )= readr:: read_tsv ("pacbio_info.tab.txt" ) %>% rename (SRASample = Sample)= sampleMeta %>% select (Sample, Pacbio) %>% rename (SRASample = Pacbio) %>% left_join (sraInfo %>% select (Run, SRASample))= sampleMeta_sel %>% group_by (Sample) %>% mutate (Run = paste0 ("-in " , Run, ".sorted.bam" )) %>% summarise (RunInputs = paste0 (Run, collapse = " " )) %>% mutate (mergeCmd = paste0 ("bamtools merge " , RunInputs, " -out " , Sample, ".sorted.bam" , " && " , " samtools index " , Sample, ".sorted.bam" ))cat (sampleMeta_sel_mergeCmd$ mergeCmd, sep = " \n " , file = "bamMergeCmds.txt" )``` ### Intersect with shared region ```{bash, eval = F} rm -f bamRegularIntersectCmds.txt && for x in bams/Pf*bai; do if [ ! -f $(basename ${x%%.sorted.bam.bai})_intersectedWithShared_chr11_chr13_region.bed ]; then echo "elucidator bamToBed --bam ${x%%.bai} | elucidator getOverlappingBedRegions --bed STDIN --intersectWithBed shared_11_13_region.bed --out $(basename ${x%%.sorted.bam.bai})_intersectedWithShared_chr11_chr13_region.bed" >> bamRegularIntersectCmds.txt; fi ; done; rm -f bamRegularMinimap2IntersectCmds.txt && for x in bamsMinimap2/Pf*bai; do if [ ! -f $(basename ${x%%.sorted.bam.bai})_minimap2_intersectedWithShared_chr11_chr13_region.bed ]; then echo "elucidator bamToBed --bam ${x%%.bai} | elucidator getOverlappingBedRegions --bed STDIN --intersectWithBed shared_11_13_region.bed --out $(basename ${x%%.sorted.bam.bai})_minimap2_intersectedWithShared_chr11_chr13_region.bed" >> bamRegularMinimap2IntersectCmds.txt; fi ; done; rm -f bamHybridIntersectCmds.txt && for x in hybridAlns/Pf*bai; do if [ ! -f $(basename ${x%%.sorted.bam.bai})_intersectedWithSharedwithHybrid_chr11_chr13_region.bed ]; then echo "elucidator bamToBed --bam ${x%%.bai} | elucidator getOverlappingBedRegions --bed STDIN --intersectWithBed combined_shared_11_13_region.bed --out $(basename ${x%%.sorted.bam.bai})_intersectedWithSharedwithHybrid_chr11_chr13_region.bed" >> bamHybridIntersectCmds.txt ; fi; done; rm -f bamHybridMinimap2IntersectCmds.txt && for x in hybridAlnsMinimap2/Pf*bai; do if [ ! -f $(basename ${x%%.sorted.bam.bai})_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region.bed ]; then echo "elucidator bamToBed --bam ${x%%.bai} | elucidator getOverlappingBedRegions --bed STDIN --intersectWithBed combined_shared_11_13_region.bed --out $(basename ${x%%.sorted.bam.bai})_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region.bed" >> bamHybridMinimap2IntersectCmds.txt; fi ; done; ``` ```{bash, eval = F} cd intersectedBeds for x in *region.bed; do if [ ! -f ${x%%.bed}_withPlotID.bed ]; then echo ${x} && elucidator bedAddSmartIDForPlotting --bed ${x} --out ${x%%.bed}_withPlotID.bed --overWrite; fi ; done; for x in *minimap2_intersectedWithShared*region.bed; do if [ ! -f ${x%%.bed}_withPlotID.bed ]; then echo ${x} && elucidator bedAddSmartIDForPlotting --bed ${x} --out ${x%%.bed}_withPlotID.bed --overWrite; fi ; done; ``` ```{r} = readr:: read_tsv ("../../../sharedBetween11_and_13/investigatingChrom11Chrom13/shared_11_13_region.bed" , col_names = F, skip = 1 )= readr:: read_tsv ("../../../sharedBetween11_and_13/investigatingChrom11Chrom13/combined_shared_11_13_region.bed" , col_names = F)``` ```{r} = tibble ()= c ("Pf3D7" ,"Pf7G8" ,"PfCD01" ,"PfDd2" ,"PfGA01" ,"PfGB4" ,"PfGN01" ,"PfHB3" ,"PfIT" ,"PfKE01" ,"PfKH01" ,"PfKH02" ,"PfML01" ,"PfSD01" ,"PfSN01" ,"PfTG01" )for (samp in samples){= paste0 ("intersectedBeds/" , samp, "_intersectedWithShared_chr11_chr13_region.bed" )if (file.exists (regionsFnp)){= readr:: read_tsv (regionsFnp, col_names = F) %>% mutate (isolate = samp)= allBwaAlns %>% bind_rows (currentAlns)= 100 = allBwaAlns %>% mutate (spanning = ifelse (== "Pf3D7_11_v3" & <= 1918029 - spanOutLimit & >= 1933390 + spanOutLimit| (== "Pf3D7_13_v3" & <= 2792022 - spanOutLimit& >= 2807397 + spanOutLimit"spanning" , "other" = allBwaAlns %>% filter (spanning == "spanning" ) %>% group_by (isolate, X1) %>% summarise (n = n ())``` ### BWA regular spanning counts ```{r} :: datatable (allBwaAlns_spanningSum %>% rename (chrom = X1),extensions = 'Buttons' , options = list (dom = 'Bfrtip' ,buttons = c ('csv' )``` ```{r} = tibble ()= c ("Pf3D7" ,"Pf7G8" ,"PfCD01" ,"PfDd2" ,"PfGA01" ,"PfGB4" ,"PfGN01" ,"PfHB3" ,"PfIT" ,"PfKE01" ,"PfKH01" ,"PfKH02" ,"PfML01" ,"PfSD01" ,"PfSN01" ,"PfTG01" )for (samp in samples){= readr:: read_tsv (paste0 ("intersectedBeds/" , samp, "_minimap2_intersectedWithShared_chr11_chr13_region_withPlotID.bed" ), col_names = F) %>% mutate (isolate = samp)= allMinimap2Alns %>% bind_rows (currentAlns)= 50 = allMinimap2Alns %>% mutate (spanning = ifelse (== "Pf3D7_11_v3" & <= 1918029 - spanOutLimit & >= 1933390 + spanOutLimit| (== "Pf3D7_13_v3" & <= 2792022 - spanOutLimit& >= 2807397 + spanOutLimit"spanning" , "other" = allMinimap2Alns %>% filter (spanning == "spanning" ) %>% group_by (isolate, X1) %>% summarise (n = n ())``` ### Minimap2 regular spanning counts ```{r} :: datatable (allMinimap2Alns_spanningSum %>% rename (chrom = X1),extensions = 'Buttons' , options = list (dom = 'Bfrtip' ,buttons = c ('csv' )``` # Plotting pacbio spanning ## Regular ```{r} = ggplot (allMinimap2Alns) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = spanning# , # color = spanning + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 30 ,ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegion+ facet_wrap (isolate~ X1, ncol = 2 , scales = "free" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" ))``` ```{r} #| fig-width: 10 #| fig-height: 30 print (allMinimap2Alns_mapPlot + facet_wrap (isolate~ X1, ncol = 2 , scales = "free" , strip.position = "right" ))``` ```{r} pdf ("allMinimap2Alns_mapPlot.pdf" , width = 20 , height = 40 , useDingbats = F)print (allMinimap2Alns_mapPlot)dev.off ()``` ```{r} = allMinimap2Alns %>% filter (isolate %in% c ("Pf3D7" , "PfCD01" , "PfHB3" , "PfSD01" ))= ggplot (allMinimap2Alns_subSetIsolates) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = spanning# , # color = spanning + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 ,ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegion+ facet_wrap (isolate~ X1, ncol = 2 , scales = "free" , strip.position = "right" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" ))``` ```{r} #| fig-width: 10 #| fig-height: 10 print (allMinimap2Alns_mapPlot_subset)``` ```{r} pdf ("allMinimap2Alns_mapPlot_subSetIsolates.pdf" , width = 20 , height = 40 , useDingbats = F)print (allMinimap2Alns_mapPlot_subset)dev.off ()``` ## Hybrid genome ```{r} = tibble ()= c ("Pf3D7" ,"Pf7G8" ,"PfCD01" ,"PfDd2" ,"PfGA01" ,"PfGB4" ,"PfGN01" ,"PfHB3" ,"PfIT" ,"PfKE01" ,"PfKH01" ,"PfKH02" ,"PfML01" ,"PfSD01" ,"PfSN01" ,"PfTG01" )for (samp in samples) {= paste0 ("intersectedBeds/" ,"_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region_withPlotID.bed" if (file.exists (regionsFnp)) {= readr:: read_tsv (regionsFnp, col_names = F) %>% mutate (isolate = samp)= allMinimap2HybridAlns %>% bind_rows (currentAlns)= 50 = allMinimap2HybridAlns %>% mutate (spanning = ifelse (== "Pf3D7_11_v3" & <= 1918029 - spanOutLimit & >= 1933390 + spanOutLimit| (== "Pf3D7_13_v3" & <= 2792022 - spanOutLimit & >= 2807397 + spanOutLimit| (== "Pf3D7_11_v3__Pf3D7_13_v3" & <= 1918029 - spanOutLimit & >= 1933404 + spanOutLimit| (== "Pf3D7_13_v3__Pf3D7_11_v3" & <= 2792022 - spanOutLimit & >= 2807383 + spanOutLimit"spanning" , "other" = allMinimap2HybridAlns %>% filter (spanning == "spanning" ) %>% group_by (isolate, X1) %>% summarise (n = n ())= ggplot (allMinimap2HybridAlns) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = spanning# , # color = spanning + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 30 ,ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegionWithHybrid+ facet_wrap (isolate~ X1, ncol = 4 , scales = "free" , strip.position = "right" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" ))``` ```{r} #| fig-width: 10 #| fig-height: 30 print (allMinimap2HybridAlns_mapPlot)``` ```{r} pdf ("allMinimap2HybridAlns_mapPlot.pdf" , width = 20 , height = 40 , useDingbats = F)print (allMinimap2HybridAlns_mapPlot)dev.off ()= allMinimap2HybridAlns %>% filter (isolate %in% c ("PfCD01" , "PfHB3" , "PfSD01" ))= ggplot (allMinimap2HybridAlns_subSetIsolates) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = spanning# , # color = spanning + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 ,ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegionWithHybrid+ facet_wrap (isolate~ X1, ncol = 4 , scales = "free" , strip.position = "right" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" ))``` ```{r} #| fig-width: 10 #| fig-height: 10 print (allMinimap2HybridAlns_mapPlot_subset)``` ```{r} pdf ("allMinimap2HybridAlns_mapPlot_subSetIsolates.pdf" , width = 20 , height = 40 , useDingbats = F)print (allMinimap2HybridAlns_mapPlot_subset)dev.off ()``` ```{r} = allMinimap2HybridAlns_subSetIsolates %>% filter ((X1 %in% c ("Pf3D7_11_v3" , "Pf3D7_13_v3" )) | == "spanning" )= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1, isolate, spanning) %>% mutate (n = n ()) %>% mutate (X8 = ifelse (spanning == "spanning" & X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ) , row_number (), X8))= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1, isolate) %>% filter (spanning == "spanning" ) %>% summarise (n = n ())= tibble ()= 50 for (isolate in unique (allMinimap2HybridAlns_subSetIsolates_filt$ isolate)){= DuplicatedRegionWithHybrid %>% filter (X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" )) %>% mutate (X2 = X2 - 10000 , X3 = X3 + 10000 )= tibble ()for (fillCount in 1 : filler_amount){= DuplicatedRegionWithHybrid_mod$ X8 = fillCount= bind_rows (currentFiler,$ isolate = isolate= bind_rows (DuplicatedRegionWithHybrid_filler, = DuplicatedRegionWithHybrid %>% select (X1, X2, X3) %>% rename (DuplicatedRegionStart = X2, DuplicatedRegionEnd = X3)= allMinimap2HybridAlns_subSetIsolates_filt %>% left_join (DuplicatedRegionWithHybrid_rename) %>% group_by (X1, X2, X3, X4) %>% mutate (X2 = max (X2, DuplicatedRegionStart - 10000 ), X3 = min (X3, DuplicatedRegionEnd + 10000 ))= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1) %>% summarise (minStart = min (X2), maxEnd = max (X3)) %>% mutate (isolate = paste0 (unique (allMinimap2HybridAlns_subSetIsolates_filt$ isolate), collapse = "," )) %>% mutate (isolate = strsplit (isolate, split = "," )) %>% unnest (isolate) %>% mutate (X8 = 1 ) %>% rename (X2 = minStart, X3 = maxEnd)= allMinimap2HybridAlns_subSetIsolates_filt %>% mutate (spanning = ifelse (%in% c ("Pf3D7_11_v3" , "Pf3D7_13_v3" ) & X2 <= DuplicatedRegionStart - 500 & X3 > DuplicatedRegionStart + 500 & X3 < DuplicatedRegionEnd), "spanFront" , %>% mutate (spanning = ifelse (%in% c ("Pf3D7_11_v3" , "Pf3D7_13_v3" ) & X2 > DuplicatedRegionStart & X3 > DuplicatedRegionEnd + 500 & X2 < DuplicatedRegionEnd - 500 ), "spanEnd" , %>% group_by (isolate, X1, spanning) %>% count () %>% group_by () %>% filter (spanning != "other" ) %>% bind_rows (tibble (isolate = "PfHB3" , X1 = "Pf3D7_11_v3__Pf3D7_13_v3" , spanning = "spanning" , n = 0 )) %>% complete (isolate, X1, spanning, fill = list (n = 0 )) %>% filter (! (grepl ("__" , X1) & spanning != "spanning" ))= allMinimap2HybridAlns_subSetIsolates_filt_spanningSum %>% group_by () %>% mutate (spanning = stringr:: str_pad (spanning, width = max (nchar (spanning)) ) ) %>% mutate (label = paste0 (spanning, "=" , n)) %>% group_by (isolate, X1) %>% summarise (label = paste0 (label, collapse = " \n " )) %>% mutate (label = ifelse (X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ), paste0 (" \n\n " , label), label))= DuplicatedRegionWithHybrid_filler %>% bind_rows (allMinimap2HybridAlns_subSetIsolates_filt_sum)= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (isolate) %>% mutate (maxPlotIDForIsolate = max (X8))= allMinimap2HybridAlns_subSetIsolates_filt %>% filter ((X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ) & == "spanning" )) %>% mutate (tenPercentMax = maxPlotIDForIsolate * 0.05 ) = ggplot (allMinimap2HybridAlns_subSetIsolates_filt %>% filter (! (X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ) & == "spanning" )) ) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = spanning# , # color = spanning + geom_rect (aes (xmin = X2, xmax = X3, ymin = 1 + (X8) * tenPercentMax, ymax = 1 + (X8 + 1 ) * tenPercentMax, fill = spanning# , # color = spanning data = allMinimap2HybridAlns_subSetIsolates_filt_hybridSpanning+ geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = "#FFFFFF00" ,data = DuplicatedRegionWithHybrid_filler+ geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 , ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegionWithHybrid+ facet_grid (isolate~ X1, scales = "free" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" ,"other" = "grey50" ,"spanning" = "#9F0162" + geom_text (mapping = aes (x = - Inf , y = - Inf , label = label),hjust = - 0.1 ,vjust = - 5 , data = allMinimap2HybridAlns_subSetIsolates_filt_spanningSum_label+ labs (x = "" , y = "" )``` ```{r} #| fig-width: 10 #| fig-height: 10 print (allMinimap2HybridAlns_mapPlot_subset_filt)``` ```{r} pdf ("allMinimap2HybridAlns_mapPlot_filt_subSetIsolates.pdf" , width = 20 , height = 15 , useDingbats = F)print (allMinimap2HybridAlns_mapPlot_subset_filt)dev.off ()``` # Plotting Nanopore spanning ```{bash, eval = F} cd intersectedBeds for x in *_nanopore_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region.bed; do echo ${x} && elucidator bedAddSmartIDForPlotting --bed ${x} --out ${x%%.bed}_withPlotID.bed --overWrite; done; ``` ```{r} = c ("Pf3D7" ,"Pf7G8" ,"PfCD01" ,"PfDd2" ,"PfGA01" ,"PfGB4" ,"PfGN01" ,"PfHB3" ,"PfIT" ,"PfKE01" ,"PfKH01" ,"PfKH02" ,"PfML01" ,"PfSD01" ,"PfSN01" ,"PfTG01" , "PfSD01PermethION" )for (samp in samples) {= paste0 ("intersectedBeds/" ,"_nanopore_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region_withPlotID.bed" = paste0 ("intersectedBeds/" ,"_nanopore_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region_filt.bed" if (file.exists (regionsFnp)) {= readr:: read_tsv (regionsFnp, col_names = F) %>% mutate (isolate = samp)= currentAlns %>% arrange (desc (X5)) %>% group_by (X4) %>% mutate (readID = row_number ()) %>% filter (readID == 1 ) %>% group_by () %>% arrange (X1, X2, X3)write_tsv (currentAlns %>% select (1 : 7 ), regionsOutFnp, col_names = F)``` ```{bash, eval = F} cd intersectedBeds for x in *_nanopore_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region_filt.bed; do echo ${x} && elucidator bedAddSmartIDForPlotting --bed ${x} --out ${x%%.bed}_withPlotID.bed --overWrite; done; ``` ```{r} = tibble ()= c ("Pf3D7" ,"Pf7G8" ,"PfCD01" ,"PfDd2" ,"PfGA01" ,"PfGB4" ,"PfGN01" ,"PfHB3" ,"PfIT" ,"PfKE01" ,"PfKH01" ,"PfKH02" ,"PfML01" ,"PfSD01" ,"PfSN01" ,"PfTG01" , "PfSD01PermethION" )for (samp in samples) {= paste0 ("intersectedBeds/" ,"_nanopore_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region_filt_withPlotID.bed" if (file.exists (regionsFnp)) {= readr:: read_tsv (regionsFnp, col_names = F) %>% mutate (isolate = samp)= allMinimap2HybridAlns %>% bind_rows (currentAlns)= 50 = allMinimap2HybridAlns %>% mutate (spanning = ifelse (== "Pf3D7_11_v3" & <= 1918029 - spanOutLimit & >= 1933390 + spanOutLimit| (== "Pf3D7_13_v3" & <= 2792022 - spanOutLimit & >= 2807397 + spanOutLimit| (== "Pf3D7_11_v3__Pf3D7_13_v3" & <= 1918029 - spanOutLimit & >= 1933404 + spanOutLimit| (== "Pf3D7_13_v3__Pf3D7_11_v3" & <= 2792022 - spanOutLimit & >= 2807383 + spanOutLimit"spanning" , "other" = allMinimap2HybridAlns %>% filter (spanning == "spanning" ) %>% group_by (isolate, X1) %>% summarise (n = n ())= ggplot (allMinimap2HybridAlns) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = spanning# , # color = spanning + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 30 ,ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegionWithHybrid+ facet_wrap (isolate~ X1, ncol = 4 , scales = "free" , strip.position = "right" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" ))pdf ("nanopore_allMinimap2HybridAlns_mapPlot.pdf" , width = 20 , height = 10 , useDingbats = F)print (allMinimap2HybridAlns_mapPlot)dev.off ()= allMinimap2HybridAlns %>% filter (isolate %in% c ("PfHB3" , "PfSD01" , "PfSD01PermethION" ))= ggplot (allMinimap2HybridAlns_subSetIsolates) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = spanning# , # color = spanning + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 ,ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegionWithHybrid+ facet_wrap (isolate~ X1, ncol = 4 , scales = "free" , strip.position = "right" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" ))``` ```{r} #| fig-width: 10 #| fig-height: 10 print (allMinimap2HybridAlns_mapPlot_subset)``` ```{r} pdf ("nanopore_allMinimap2HybridAlns_mapPlot_subSetIsolates.pdf" , width = 20 , height = 10 , useDingbats = F)print (allMinimap2HybridAlns_mapPlot_subset)dev.off ()``` ```{r} = allMinimap2HybridAlns_subSetIsolates %>% filter ((X1 %in% c ("Pf3D7_11_v3" , "Pf3D7_13_v3" )) | == "spanning" )= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1, isolate, spanning) %>% mutate (n = n ()) %>% mutate (X8 = ifelse (spanning == "spanning" & X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ) , row_number (), X8))= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1, isolate) %>% filter (spanning == "spanning" ) %>% summarise (n = n ())= tibble ()= 50 for (isolate in unique (allMinimap2HybridAlns_subSetIsolates_filt$ isolate)){= DuplicatedRegionWithHybrid %>% filter (X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" )) %>% mutate (X2 = X2 - 10000 , X3 = X3 + 10000 )= tibble ()for (fillCount in 1 : filler_amount){= DuplicatedRegionWithHybrid_mod$ X8 = fillCount= bind_rows (currentFiler,$ isolate = isolate= bind_rows (DuplicatedRegionWithHybrid_filler, = DuplicatedRegionWithHybrid %>% select (X1, X2, X3) %>% rename (DuplicatedRegionStart = X2, DuplicatedRegionEnd = X3)= allMinimap2HybridAlns_subSetIsolates_filt %>% left_join (DuplicatedRegionWithHybrid_rename) %>% group_by (X1, X2, X3, X4) %>% mutate (X2 = max (X2, DuplicatedRegionStart - 10000 ), X3 = min (X3, DuplicatedRegionEnd + 10000 ))= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1) %>% summarise (minStart = min (X2), maxEnd = max (X3)) %>% mutate (isolate = paste0 (unique (allMinimap2HybridAlns_subSetIsolates_filt$ isolate), collapse = "," )) %>% mutate (isolate = strsplit (isolate, split = "," )) %>% unnest (isolate) %>% mutate (X8 = 1 ) %>% rename (X2 = minStart, X3 = maxEnd)= allMinimap2HybridAlns_subSetIsolates_filt %>% mutate (spanning = ifelse (%in% c ("Pf3D7_11_v3" , "Pf3D7_13_v3" ) & X2 <= DuplicatedRegionStart - 500 & X3 > DuplicatedRegionStart + 500 & X3 < DuplicatedRegionEnd), "spanFront" , %>% mutate (spanning = ifelse (%in% c ("Pf3D7_11_v3" , "Pf3D7_13_v3" ) & X2 > DuplicatedRegionStart & X3 > DuplicatedRegionEnd + 500 & X2 < DuplicatedRegionEnd - 500 ), "spanEnd" , %>% group_by (isolate, X1, spanning) %>% count () %>% group_by () %>% filter (spanning != "other" ) %>% bind_rows (tibble (isolate = "PfHB3" , X1 = "Pf3D7_11_v3__Pf3D7_13_v3" , spanning = "spanning" , n = 0 )) %>% complete (isolate, X1, spanning, fill = list (n = 0 )) %>% filter (! (grepl ("__" , X1) & spanning != "spanning" ))= allMinimap2HybridAlns_subSetIsolates_filt_spanningSum %>% group_by () %>% mutate (spanning = stringr:: str_pad (spanning, width = max (nchar (spanning)) ) ) %>% mutate (label = paste0 (spanning, "=" , n)) %>% group_by (isolate, X1) %>% summarise (label = paste0 (label, collapse = " \n " )) %>% mutate (label = ifelse (X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ), paste0 (" \n\n " , label), label))= DuplicatedRegionWithHybrid_filler %>% bind_rows (allMinimap2HybridAlns_subSetIsolates_filt_sum)= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (isolate) %>% mutate (maxPlotIDForIsolate = max (X8))= allMinimap2HybridAlns_subSetIsolates_filt %>% filter ((X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ) & == "spanning" )) %>% mutate (subPercent = maxPlotIDForIsolate * 0.01 ) = ggplot (allMinimap2HybridAlns_subSetIsolates_filt %>% filter (! (X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ) & == "spanning" )) ) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = spanning# , # color = spanning + geom_rect (aes (xmin = X2, xmax = X3, ymin = 1 + (X8) * subPercent, ymax = 1 + (X8 + 1 ) * subPercent, fill = spanning# , # color = spanning data = allMinimap2HybridAlns_subSetIsolates_filt_hybridSpanning+ geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = "#FFFFFF00" ,data = DuplicatedRegionWithHybrid_filler+ geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 , ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegionWithHybrid+ facet_grid (isolate~ X1, scales = "free" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" ,"other" = "grey50" ,"spanning" = "#9F0162" + geom_text (mapping = aes (x = - Inf , y = - Inf , label = label),hjust = - 0.1 ,vjust = - 5 , data = allMinimap2HybridAlns_subSetIsolates_filt_spanningSum_label+ labs (x = "" , y = "" )``` ```{r} #| fig-width: 10 #| fig-height: 10 print (allMinimap2HybridAlns_mapPlot_subset_filt)``` ```{r} pdf ("nanopore_allMinimap2HybridAlns_mapPlot_filt_subSetIsolates.pdf" , width = 20 , height = 10 , useDingbats = F)print (allMinimap2HybridAlns_mapPlot_subset_filt)dev.off ()``` # Plotting by combining pacbio and nanopore reads ```{r} = c ("Pf3D7" ,"Pf7G8" ,"PfCD01" ,"PfDd2" ,"PfGA01" ,"PfGB4" ,"PfGN01" ,"PfHB3" ,"PfIT" ,"PfKE01" ,"PfKH01" ,"PfKH02" ,"PfML01" ,"PfSD01" ,"PfSN01" ,"PfTG01" )for (samp in samples) {= tibble ()= paste0 ("intersectedBeds/" ,"_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region_filt_withPlotID.bed" if (file.exists (regionsFnp)) {= readr:: read_tsv (regionsFnp, col_names = F) %>% mutate (isolate = samp)= currentAlnsForSamp %>% bind_rows (currentAlns)= paste0 ("intersectedBeds/" ,"_nanopore_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region_filt_withPlotID.bed" = paste0 ("intersectedBeds/" ,"PermethION" ,"_nanopore_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region_filt_withPlotID.bed" if (file.exists (permethion_nanoRegionsFnp)) {= readr:: read_tsv (permethion_nanoRegionsFnp, col_names = F) %>% mutate (isolate = samp) # %>% # mutate(X8 = X8 + maxX8) = currentAlnsForSamp %>% bind_rows (currentAlns)else if (file.exists (nanoRegionsFnp)) {= readr:: read_tsv (nanoRegionsFnp, col_names = F) %>% mutate (isolate = samp) # %>% # mutate(X8 = X8 + maxX8) = currentAlnsForSamp %>% bind_rows (currentAlns)if (file.exists (regionsFnp)) {= currentAlnsForSamp %>% arrange (desc (X5)) %>% group_by () %>% arrange (X1, X2, X3)= paste0 ("intersectedBeds/" ,"_combined_intersectedWithSharedwithHybrid_chr11_chr13_region_filt.bed" write_tsv (currentAlnsForSamp %>% select (1 : 7 ), regionsOutFnp, col_names = F)``` ```{bash, eval = F} cd intersectedBeds rm -f runAddPlotID_to_combined_intersectedWithSharedwithHybrid_chr11_chr13_regionCmds.txt && for x in *_combined_intersectedWithSharedwithHybrid_chr11_chr13_region_filt.bed; do echo "elucidator bedAddSmartIDForPlotting --bed ${x} --out ${x%%.bed}_withPlotID.bed --overWrite " >> runAddPlotID_to_combined_intersectedWithSharedwithHybrid_chr11_chr13_regionCmds.txt ; done; elucidator runMultipleCommands --cmdFile runAddPlotID_to_combined_intersectedWithSharedwithHybrid_chr11_chr13_regionCmds.txt --numThreads 15 --raw ``` ```{bash, eval = F} cd intersectedBeds for x in *_combined_intersectedWithSharedwithHybrid_chr11_chr13_region.bed; do elucidator mergeOverlappingRegionsSameName --bedFnp ${x} --overWrite --out ${x%%_combined_intersectedWithSharedwithHybrid_chr11_chr13_region.bed}_mergedCombinedFinalReads.bed ; done; rm -f runAddPlotID_to_mergedCombinedFinalReadsCmds.txt && for x in *_mergedCombinedFinalReads.bed; do echo "elucidator bedAddSmartIDForPlotting --bed ${x} --out ${x%%.bed}_withPlotID.bed --overWrite " >> runAddPlotID_to_mergedCombinedFinalReadsCmds.txt ; done; elucidator runMultipleCommands --cmdFile runAddPlotID_to_mergedCombinedFinalReadsCmds.txt --numThreads 15 --raw ``` ```{r} = tibble ()= c ("Pf3D7" ,"Pf7G8" ,"PfCD01" ,"PfDd2" ,"PfGA01" ,"PfGB4" ,"PfGN01" ,"PfHB3" ,"PfIT" ,"PfKE01" ,"PfKH01" ,"PfKH02" ,"PfML01" ,"PfSD01" ,"PfSN01" ,"PfTG01" )for (samp in samples) {= paste0 ("intersectedBeds/" ,"_mergedCombinedFinalReads_withPlotID.bed" if (file.exists (regionsFnp)) {= readr:: read_tsv (regionsFnp, col_names = F) %>% mutate (isolate = samp)= allMinimap2HybridAlns %>% bind_rows (currentAlns)= 50 = allMinimap2HybridAlns %>% mutate (spanning = ifelse (== "Pf3D7_11_v3" & <= 1918029 - spanOutLimit & >= 1933390 + spanOutLimit| (== "Pf3D7_13_v3" & <= 2792022 - spanOutLimit & >= 2807397 + spanOutLimit| (== "Pf3D7_11_v3__Pf3D7_13_v3" & <= 1918029 - spanOutLimit & >= 1933404 + spanOutLimit| (== "Pf3D7_13_v3__Pf3D7_11_v3" & <= 2792022 - spanOutLimit & >= 2807383 + spanOutLimit"spanning" , "other" = allMinimap2HybridAlns %>% filter (spanning == "spanning" ) %>% group_by (isolate, X1) %>% summarise (n = n ())= ggplot (allMinimap2HybridAlns) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = spanning# , # color = spanning + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 30 ,ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegionWithHybrid+ facet_wrap (isolate~ X1, ncol = 4 , scales = "free" , strip.position = "right" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" ))pdf ("combined_allMinimap2HybridAlns_mapPlot.pdf" , width = 20 , height = 40 , useDingbats = F)print (allMinimap2HybridAlns_mapPlot)dev.off ()= allMinimap2HybridAlns %>% filter (isolate %in% c ("PfCD01" , "PfHB3" , "PfSD01" ))= ggplot (allMinimap2HybridAlns_subSetIsolates) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = spanning# , # color = spanning + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 ,ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegionWithHybrid+ facet_wrap (isolate~ X1, ncol = 4 , scales = "free" , strip.position = "right" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" ))pdf ("combined_allMinimap2HybridAlns_mapPlot_subSetIsolates.pdf" , width = 20 , height = 40 , useDingbats = F)print (allMinimap2HybridAlns_mapPlot_subset)dev.off ()``` ```{r} = allMinimap2HybridAlns_subSetIsolates %>% filter ((X1 %in% c ("Pf3D7_11_v3" , "Pf3D7_13_v3" )) | == "spanning" )= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1, isolate, spanning) %>% mutate (n = n ()) %>% mutate (X8 = ifelse (spanning == "spanning" & X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ) , row_number (), X8))= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1, isolate) %>% filter (spanning == "spanning" ) %>% summarise (n = n ())= tibble ()= 50 for (isolate in unique (allMinimap2HybridAlns_subSetIsolates_filt$ isolate)){= DuplicatedRegionWithHybrid %>% filter (X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" )) %>% mutate (X2 = X2 - 10000 , X3 = X3 + 10000 )= tibble ()for (fillCount in 1 : filler_amount){= DuplicatedRegionWithHybrid_mod$ X8 = fillCount= bind_rows (currentFiler,$ isolate = isolate= bind_rows (DuplicatedRegionWithHybrid_filler, = DuplicatedRegionWithHybrid %>% select (X1, X2, X3) %>% rename (DuplicatedRegionStart = X2, DuplicatedRegionEnd = X3)= allMinimap2HybridAlns_subSetIsolates_filt %>% left_join (DuplicatedRegionWithHybrid_rename) %>% group_by (X1, X2, X3, X4) %>% mutate (X2 = max (X2, DuplicatedRegionStart - 10000 ), X3 = min (X3, DuplicatedRegionEnd + 10000 ))= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1) %>% summarise (minStart = min (X2), maxEnd = max (X3)) %>% mutate (isolate = paste0 (unique (allMinimap2HybridAlns_subSetIsolates_filt$ isolate), collapse = "," )) %>% mutate (isolate = strsplit (isolate, split = "," )) %>% unnest (isolate) %>% mutate (X8 = 1 ) %>% rename (X2 = minStart, X3 = maxEnd)= allMinimap2HybridAlns_subSetIsolates_filt %>% mutate (spanning = ifelse (%in% c ("Pf3D7_11_v3" , "Pf3D7_13_v3" ) & X2 <= DuplicatedRegionStart - 500 & X3 > DuplicatedRegionStart + 500 & X3 < DuplicatedRegionEnd), "spanFront" , %>% mutate (spanning = ifelse (%in% c ("Pf3D7_11_v3" , "Pf3D7_13_v3" ) & X2 > DuplicatedRegionStart & X3 > DuplicatedRegionEnd + 500 & X2 < DuplicatedRegionEnd - 500 ), "spanEnd" , %>% group_by (isolate, X1, spanning) %>% count () %>% group_by () %>% filter (spanning != "other" ) %>% bind_rows (tibble (isolate = "PfHB3" , X1 = "Pf3D7_11_v3__Pf3D7_13_v3" , spanning = "spanning" , n = 0 )) %>% complete (isolate, X1, spanning, fill = list (n = 0 )) %>% filter (! (grepl ("__" , X1) & spanning != "spanning" ))= allMinimap2HybridAlns_subSetIsolates_filt_spanningSum %>% group_by () %>% mutate (spanning = stringr:: str_pad (spanning, width = max (nchar (spanning)) ) ) %>% mutate (label = paste0 (spanning, "=" , n)) %>% group_by (isolate, X1) %>% summarise (label = paste0 (label, collapse = " \n " )) %>% mutate (label = ifelse (X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ), paste0 (" \n\n " , label), label))= DuplicatedRegionWithHybrid_filler %>% bind_rows (allMinimap2HybridAlns_subSetIsolates_filt_sum)= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (isolate) %>% mutate (maxPlotIDForIsolate = max (X8))= allMinimap2HybridAlns_subSetIsolates_filt %>% filter ((X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ) & == "spanning" )) %>% mutate (subPercent = maxPlotIDForIsolate * 0.01 ) = ggplot (allMinimap2HybridAlns_subSetIsolates_filt %>% filter (! (X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" ) & == "spanning" )) ) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = spanning# , # color = spanning + geom_rect (aes (xmin = X2, xmax = X3, ymin = 1 + (X8) * subPercent, ymax = 1 + (X8 + 1 ) * subPercent, fill = spanning# , # color = spanning data = allMinimap2HybridAlns_subSetIsolates_filt_hybridSpanning+ geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = "#FFFFFF00" ,data = DuplicatedRegionWithHybrid_filler+ geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 , ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegionWithHybrid+ facet_grid (isolate~ X1, scales = "free" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" ,"other" = "grey50" ,"spanning" = "#9F0162" + geom_text (mapping = aes (x = - Inf , y = - Inf , label = label),hjust = - 0.1 ,vjust = - 3.4 , data = allMinimap2HybridAlns_subSetIsolates_filt_spanningSum_label+ labs (x = "" , y = "" )pdf ("combined_allMinimap2HybridAlns_mapPlot_filt_subSetIsolates.pdf" , width = 20 , height = 10 , useDingbats = F)print (allMinimap2HybridAlns_mapPlot_subset_filt)dev.off ()pdf ("spanning_reads_across_PfCD01_PfHB3_PfSD01.pdf" , width = 20 , height = 10 , useDingbats = F)print (allMinimap2HybridAlns_mapPlot_subset_filt)dev.off ()``` ```{r} #| fig-column: screen #| fig-width: 20 #| fig-height: 10 print (allMinimap2HybridAlns_mapPlot_subset_filt)``` ```{r} = allMinimap2HybridAlns_subSetIsolates= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1, isolate, spanning) %>% mutate (n = n ())= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1, isolate) %>% filter (spanning == "spanning" ) %>% summarise (n = n ())= tibble ()= 50 for (isolate in unique (allMinimap2HybridAlns_subSetIsolates_filt$ isolate)){= DuplicatedRegionWithHybrid %>% filter (X1 %in% c ("Pf3D7_13_v3__Pf3D7_11_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" )) %>% mutate (X2 = X2 - 10000 , X3 = X3 + 10000 )= tibble ()for (fillCount in 1 : filler_amount){= DuplicatedRegionWithHybrid_mod$ X8 = fillCount= bind_rows (currentFiler,$ isolate = isolate= bind_rows (DuplicatedRegionWithHybrid_filler, = DuplicatedRegionWithHybrid %>% select (X1, X2, X3) %>% rename (DuplicatedRegionStart = X2, DuplicatedRegionEnd = X3)= allMinimap2HybridAlns_subSetIsolates_filt %>% left_join (DuplicatedRegionWithHybrid_rename) %>% group_by (X1, X2, X3, X4) %>% mutate (X2 = max (X2, DuplicatedRegionStart - 10000 ), X3 = min (X3, DuplicatedRegionEnd + 10000 ))= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (X1) %>% summarise (minStart = min (X2), maxEnd = max (X3)) %>% mutate (isolate = paste0 (unique (allMinimap2HybridAlns_subSetIsolates_filt$ isolate), collapse = "," )) %>% mutate (isolate = strsplit (isolate, split = "," )) %>% unnest (isolate) %>% mutate (X8 = 1 ) %>% rename (X2 = minStart, X3 = maxEnd)= allMinimap2HybridAlns_subSetIsolates_filt %>% mutate (spanning = ifelse (<= DuplicatedRegionStart - 500 & X3 > DuplicatedRegionStart + 500 & X3 < DuplicatedRegionEnd), "spanFront" , %>% mutate (spanning = ifelse (> DuplicatedRegionStart & X3 > DuplicatedRegionEnd + 500 & X2 < DuplicatedRegionEnd - 500 ), "spanEnd" , %>% group_by (isolate, X1, spanning) %>% count () %>% group_by () %>% filter (spanning != "other" ) %>% bind_rows (tibble (isolate = "PfHB3" , X1 = "Pf3D7_11_v3__Pf3D7_13_v3" , spanning = "spanning" , n = 0 )) %>% complete (isolate, X1, spanning, fill = list (n = 0 ))= allMinimap2HybridAlns_subSetIsolates_filt_spanningSum %>% group_by () %>% mutate (spanning = stringr:: str_pad (spanning, width = max (nchar (spanning)) ) ) %>% mutate (label = paste0 (spanning, "=" , n)) %>% group_by (isolate, X1) %>% summarise (label = paste0 (label, collapse = " \n " )) = DuplicatedRegionWithHybrid_filler %>% bind_rows (allMinimap2HybridAlns_subSetIsolates_filt_sum)= allMinimap2HybridAlns_subSetIsolates_filt %>% group_by (isolate) %>% mutate (maxPlotIDForIsolate = max (X8))``` ```{r} = ggplot () + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8, ymax = X8 + 1 , fill = spanning# , # color = spanning data = allMinimap2HybridAlns_subSetIsolates_filt# data = allMinimap2HybridAlns_subSetIsolates_filt %>% # filter(!( # X1 %in% c("Pf3D7_13_v3__Pf3D7_11_v3", "Pf3D7_11_v3__Pf3D7_13_v3") & # spanning == "spanning" # )) + # geom_rect(aes(xmin = X2, xmax = X3, # ymin = 1 + (X8) * subPercent, ymax = 1 + (X8 + 1) * subPercent, # fill = spanning # # , # # color = spanning # ), # data = allMinimap2HybridAlns_subSetIsolates_filt_hybridSpanning # ) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8,ymax = X8 + 1 , fill = "#FFFFFF00" ,data = DuplicatedRegionWithHybrid_filler+ geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 , ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegionWithHybrid+ facet_grid (isolate~ X1, scales = "free" ) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" ,"other" = "grey50" ,"spanning" = "#9F0162" guide = "none" ) + geom_text (mapping = aes (x = - Inf , y = - Inf , label = label),hjust = - 0.1 / 1.6 ,vjust = - 3.4 / 1.6 , data = allMinimap2HybridAlns_subSetIsolates_filt_spanningSum_label+ labs (x = "" , y = "" , fill = "" )``` ```{r} pdf ("spanning_reads_across_allReads_PfCD01_PfHB3_PfSD01.pdf" , width = 20 / 1.4 , height = 10 / 1.4 , useDingbats = F,family = "mono" )print (allMinimap2HybridAlns_mapPlot_subset_filt)dev.off ()``` ```{r} #| fig-column: screen #| fig-width: 20 #| fig-height: 10 print (allMinimap2HybridAlns_mapPlot_subset_filt)``` ```{r} = allMinimap2HybridAlns_subSetIsolates_filt %>% filter (X5 > 15000 ) %>% group_by (X1, X2, X3, isolate) %>% arrange (desc (spanning)) for (iso in unique (allMinimap2HybridAlns_subSetIsolates_filt_filt$ isolate)){= allMinimap2HybridAlns_subSetIsolates_filt_filt %>% filter (iso == isolate)write_tsv (allMinimap2HybridAlns_subSetIsolates_filt_filt_iso, col_names = F, file = paste0 (iso, "_allMinimap2HybridAlns_subSetIsolates_filt_filt.bed" ))``` ```{bash, eval = T} elucidator bedAddSmartIDForPlotting --bed PfCD01_allMinimap2HybridAlns_subSetIsolates_filt_filt.bed --overWrite --out PfCD01_allMinimap2HybridAlns_subSetIsolates_filt_filt_withID.bed elucidator bedAddSmartIDForPlotting --bed PfSD01_allMinimap2HybridAlns_subSetIsolates_filt_filt.bed --overWrite --out PfSD01_allMinimap2HybridAlns_subSetIsolates_filt_filt_withID.bed elucidator bedAddSmartIDForPlotting --bed PfHB3_allMinimap2HybridAlns_subSetIsolates_filt_filt.bed --overWrite --out PfHB3_allMinimap2HybridAlns_subSetIsolates_filt_filt_withID.bed ``` ```{r} = tibble ()for (iso in unique (allMinimap2HybridAlns_subSetIsolates_filt_filt$ isolate)){= readr:: read_tsv (paste0 (iso, "_allMinimap2HybridAlns_subSetIsolates_filt_filt_withID.bed" ), col_names = F)= bind_rows (allMinimap2HybridAlns_subSetIsolates_filt_filt_withIds, colnames (allMinimap2HybridAlns_subSetIsolates_filt_filt_withIds) = c (colnames (allMinimap2HybridAlns_subSetIsolates_filt_filt), "plotID" )``` ```{r} = allMinimap2HybridAlns_subSetIsolates_filt_spanningSum_label %>% mutate (label = gsub (".*spanning" , "spanning" , label))<- c ("3D7 Chr11" , "3D7 Chr13" , "Hybrid 3D7 Chr11-13" , "Hybrid 3D7 Chr13-11" )names (ChromLabels) <- c ("Pf3D7_11_v3" , "Pf3D7_13_v3" , "Pf3D7_11_v3__Pf3D7_13_v3" , "Pf3D7_13_v3__Pf3D7_11_v3" )= ggplot () + geom_rect (aes (xmin = X2, xmax = X3, ymin = plotID, ymax = plotID + 1 , fill = spanning# , # color = spanning data = allMinimap2HybridAlns_subSetIsolates_filt_filt_withIds# data = allMinimap2HybridAlns_subSetIsolates_filt %>% # filter(!( # X1 %in% c("Pf3D7_13_v3__Pf3D7_11_v3", "Pf3D7_11_v3__Pf3D7_13_v3") & # spanning == "spanning" # )) + # geom_rect(aes(xmin = X2, xmax = X3, # ymin = 1 + (X8) * subPercent, ymax = 1 + (X8 + 1) * subPercent, # fill = spanning # # , # # color = spanning # ), # data = allMinimap2HybridAlns_subSetIsolates_filt_hybridSpanning # ) + geom_rect (aes (xmin = X2, xmax = X3, ymin = X8, ymax = X8 + 1 , fill = "#FFFFFF00" ,data = DuplicatedRegionWithHybrid_filler+ geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 , ymax = 0 , fill = "DuplicatedRegion" ), data = DuplicatedRegionWithHybrid+ facet_grid (isolate~ X1, scales = "free" , labeller = labeller (X1 = ChromLabels)) + + theme (axis.text.x = element_text (size= 12 , angle = - 45 , vjust = 0.5 , hjust = 0 )) + scale_fill_manual (values = c ("DuplicatedRegion" = "#2271B2" , "other" = "grey50" , "spanning" = "#9F0162" ), labels = c ("Duplicated Region" , "Non-spanning Reads" , "Spanning Reads" )) + scale_color_manual (values = c ("DuplicatedRegion" = "#2271B2" ,"other" = "grey50" ,"spanning" = "#9F0162" labels = c ("Duplicated Region" , "Non-spanning Reads" , "Spanning Reads" ), guide = "none" ) + geom_text (mapping = aes (x = - Inf , y = - Inf , label = label),hjust = - 0.25 ,vjust = - 12.5 + 2 , data = allMinimap2HybridAlns_subSetIsolates_filt_spanningSum_label_forFiltsize = 5 ,+ labs (x = "" , y = "" , fill = "" ) + theme (strip.text.x = element_text (size = 20 ), strip.text.y = element_text (size = 20 ))+ theme (legend.text = element_text (size = 20 ), legend.title = element_text (size = 20 , face = "bold" ), legend.box= "vertical" , legend.margin= margin (),legend.background = element_blank ()) ``` ```{r} pdf ("filt_spanning_reads_across_allReads_PfCD01_PfHB3_PfSD01.pdf" , width = 20 / 1.4 , height = 10 / 1.4 , useDingbats = F)print (allMinimap2HybridAlns_mapPlot_subset_filt_filt)dev.off ()``` ```{r} #| fig-column: screen #| fig-width: 20 #| fig-height: 10 print (allMinimap2HybridAlns_mapPlot_subset_filt_filt)``` ```{r,echo=F, eval=F} sd01_nano = readr::read_tsv("intersectedBeds/PfSD01_nanopore_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region.bed", col_names = F) sd01_permethion = readr::read_tsv("intersectedBeds/PfSD01_nanopore_minimap2_intersectedWithSharedwithHybrid_chr11_chr13_region.bed", col_names = F) sd01_nano_filt = sd01_nano %>% filter(X4 %!in% sd01_permethion$X4) ```