Code
nonParalogousStableWindows_all = readr::read_tsv("windows/mergedFinalLargeWindows.bed", col_names = F)
create_dt(nonParalogousStableWindows_all)In order to study genomic deletions in the unstable and frequent recombinant sub/peri-telomeric regions surrounding HRP2 and HRP3 and related regions, ideal regions surrounding these genes were determined. Ideal genomic regions were created by selecting regions that were conserved between strains and did not contain paralogous regions in order to best determine where possible deletions were occurring.
This was done by
The merged windows from the above analysis.
nonParalogousStableWindows_all = readr::read_tsv("windows/mergedFinalLargeWindows.bed", col_names = F)
create_dt(nonParalogousStableWindows_all)The regions investigated for pfhrp2 and pfhrp3 deletions.
finalHRPII_HRPIII_windows_withTuned = readr::read_tsv("windows/finalHRPII_HRPIII_windows_withTuned.bed", col_names = F) %>%
mutate(genomicID = paste0(X1, "-", X2, "-", X3)) %>%
mutate(chrom = X1, len = X3 -X2) %>%
rename(name = X4)
create_dt(finalHRPII_HRPIII_windows_withTuned %>%
select(-chrom, -len) %>%
rename(chrom= X1, start = X2, end = X3, length = X5, strand = X6, geneInfo = X7))Local haplotype reconstruction was performed on each region for over 16,000 whole genome sequences (WGS) publicly available P. falciparum field samples. The called haplotypes were analyzed to determine which subregions contained variation in order to genotype each chromosome.
finalHRPII_HRPIII_windows_subWindows_withTuned = readr::read_tsv("windows/finalHRPII_HRPIII_windows_withTuned_combinedVarConservedRegions.bed", col_names = F) %>%
mutate(genomicID = paste0(X1, "-", X2, "-", X3)) %>%
mutate(chrom = X1, len = X3 - X2) %>%
rename(name = X4)
create_dt(finalHRPII_HRPIII_windows_subWindows_withTuned %>%
select(-chrom, -len) %>%
rename(chrom= X1, start = X2, end = X3, length = X5, strand = X6, geneInfo = X7))gatk_calls_hrp2_3Regions_database_hrpsallMetaDeletionCalls = readr::read_tsv("../DeletionPatternAnalysis/wgs_variants/THEREALMcCOIL/categorical_method/split_filtered_gatk_calls_hrp2-3Regions_database_hrpsallMetaDeletionCalls.bed", col_names = T) %>%
rename(chrom = `#chrom`)Reading in gene annotation for the regions of interest
chroms = readr::read_tsv("../MappingOutSurroundingRegions/surroundingRegionsMaterials/chromLengths/Pf3D7.txt", col_names = c("chrom", "length"))
chrom08_genes = readr::read_tsv("../MappingOutSurroundingRegions/surroundingRegionsMaterials/endBeds/split_Pf3D7_chrom08_toEnd_genes.tab.txt", col_names = T)
chrom11_genes = readr::read_tsv("../MappingOutSurroundingRegions/surroundingRegionsMaterials/endBeds/split_Pf3D7_chrom11_toEnd_genes.tab.txt", col_names = T)
chrom13_genes = readr::read_tsv("../MappingOutSurroundingRegions/surroundingRegionsMaterials/endBeds/split_Pf3D7_chrom13_toEnd_genes.tab.txt", col_names = T)
allGenes = bind_rows(chrom08_genes, chrom11_genes, chrom13_genes) %>%
rename(chrom = col.0, start = col.1, end = col.2, name = col.3, length = col.4, strand = col.5) %>%
mutate(rawGeneDescription = description) %>%
mutate(gene = ifelse(grepl("PF3D7_0831800", description), "HRP II", "other")) %>%
mutate(gene = ifelse(grepl("PF3D7_1372200", description), "HRP III", gene)) %>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse("membrane associated erythrocyte binding-like protein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>%
mutate(description = ifelse("membrane associated erythrocyte binding-likeprotein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>%
mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description)) %>%
mutate(description = gsub(",putative", ", putative", description)) %>%
mutate(description = gsub(",pseudogene", ", pseudogene", description))%>%
mutate(description = gsub("unknownfunction", "unknown function", description))%>%
mutate(description = gsub("conserved protein, unknown function", "conserved Plasmodium protein, unknown function", description)) %>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description))%>%
mutate(description = gsub(",pseudogene", ", pseudogene", description))%>%
mutate(description = gsub("unknownfunction", "unknown function", description))%>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse(grepl("sporozoite and liver stage tryptophan-rich protein, putative", description), "tryptophan/threonine-rich antigen", description))%>%
mutate(description = ifelse(grepl("CRA domain-containing protein, putative", description), "conserved Plasmodium protein, unknown function", description))%>%
mutate(description = gsub(",pseudogene", ", pseudogene", description)) %>%
mutate(description = gsub("surfaceantigen", "surface antigen", description)) %>%
mutate(description = gsub("Tetratricopeptide repeat, putative", "tetratricopeptide repeat protein, putative", description)) %>%
mutate(description = gsub("transmembraneprotein", "transmembrane protein", description)) %>%
mutate(description = ifelse(grepl("PfEMP1", description) & grepl("pseudogene", description), "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>%
mutate(description = gsub("PIR protein", "stevor", description)) %>%
mutate(description = gsub("erythrocyte membrane protein 1-like", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>%
mutate(description = gsub("acidic terminal segments, variant surface antigen of PfEMP1, putative", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description))%>%
mutate(description = ifelse(grepl("CoA binding protein", description, ignore.case = T), "acyl-CoA binding protein", description)) %>%
mutate(description = ifelse(grepl("transfer RNA", description) | grepl("tRNA", description), "tRNA", description))%>%
mutate(description = ifelse(grepl("cytoadherence", description), "CLAG", description))%>%
mutate(description = ifelse(grepl("surface-associated interspersed protein", description), "SURFIN", description))%>%
mutate(description = ifelse(grepl("SURFIN", description), "SURFIN", description))%>%
mutate(description = ifelse(grepl("stevor-like", description), "stevor, pseudogene", description)) %>%
mutate(description = ifelse(grepl("exported protein family", description), "exported protein family", description)) %>%
mutate(description = ifelse(grepl("ribosomal RNA", description), "rRNA", description)) %>%
mutate(description = ifelse(grepl("serine/threonine protein kinase", description), "serine/threonine protein kinase, FIKK family", description)) %>%
mutate(description = ifelse(grepl("hypothetical protein", description), "hypothetical protein, conserved", description)) %>%
mutate(description = ifelse(grepl("conserved Plasmodium protein, unknown function", description), "hypothetical protein, conserved", description)) %>%
mutate(description = ifelse(grepl("Rifin/stevor family, putative", description), "stevor", description)) %>%
mutate(description = ifelse(grepl("stevor", description), "stevor", description)) %>%
mutate(description = ifelse(grepl("rifin", description), "rifin", description)) %>%
mutate(description = ifelse(grepl("erythrocyte membrane protein 1 (PfEMP1), pseudogene", description), "erythrocyte membrane protein 1 (PfEMP1)", description)) %>%
mutate(description = ifelse(grepl("erythrocyte membrane protein 1", description), "erythrocyte membrane protein 1 (PfEMP1)", description)) %>%
mutate(description = ifelse(grepl("probably protein", description), "unspecified product", description)) %>%
mutate(description = ifelse(grepl("RESA", description), "RESA", description)) %>%
mutate(description = ifelse(grepl("ring-infected erythrocyte surface antigen", description), "ring-infected erythrocyte surface antigen", description))%>%
mutate(description = ifelse(grepl("Duffy binding domain/Erythrocyte binding antigen175, putative", description), "erythrocyte binding like protein 1", description)) %>%
mutate(description = ifelse(grepl("erythrocyte binding like protein 1", description), "erythrocyte binding like protein 1", description))%>%
mutate(description = ifelse(grepl("unspecified product", description), "hypothetical protein, conserved", description))%>%
mutate(description = ifelse(grepl("probable protein, unknown function", description), "hypothetical protein, conserved", description)) %>%
mutate(description = ifelse(grepl("RESA", description), "ring-infected erythrocyte surface antigen", description))
descriptionColorsNames = unique(c(allGenes$description))
descriptionColors = scheme$hex(length(descriptionColorsNames))
names(descriptionColors) = descriptionColorsNames
allGenes_hrps = allGenes %>%
filter(grepl("histidine-rich protein II", description))finalHRPII_HRPIII_windows_withTuned_sum = finalHRPII_HRPIII_windows_withTuned %>%
group_by(chrom) %>%
summarise(minStart = min(X2),
maxEnd = max(X3))
shared_chrom11_chrom13 = readr::read_tsv("../rRNA_segmental_duplications/sharedBetween11_and_13/investigatingChrom11Chrom13/shared_11_13_region.bed") %>%
rename(chrom = `#chrom`)
transitionPointWithChr5Dup = tibble(
chrom = "Pf3D7_13_v3",
start = 2835587,
end = 2835587 + 1,
name = "TransitionPointWithChr5Dup",
len = 1,
strand = '-'
)
tare = readr::read_tsv("../MappingOutSurroundingRegions/surroundingRegionsMaterials/alltrfs/trf_Pf3D7/allRepeats_filt_starts_ends_tares.tsv")
tare_filt = tare %>%
filter(X1 %in% finalHRPII_HRPIII_windows_withTuned_sum$chrom) %>%
left_join(finalHRPII_HRPIII_windows_withTuned_sum %>%
rename(X1 = chrom)) %>%
filter(X2 >=minStart) %>%
mutate(repeatType = gsub("TARE1", "Telomere associated Repetitive Element 1", repeatType))%>%
mutate(repeatType = gsub("SB3", "Telomere associated Repetitive Element 3", repeatType))
variants_filt = gatk_calls_hrp2_3Regions_database_hrpsallMetaDeletionCalls %>%
filter(chrom %in% finalHRPII_HRPIII_windows_withTuned_sum$chrom) %>%
left_join(finalHRPII_HRPIII_windows_withTuned_sum) %>%
filter(start >=minStart)
chroms_filt = chroms %>%
left_join(finalHRPII_HRPIII_windows_withTuned_sum) %>%
filter(chrom %in% finalHRPII_HRPIII_windows_withTuned$X1)
blank_chroms = chroms_filt%>%
rename(end = length) %>%
mutate(length = end - minStart) %>%
mutate(newEnd = minStart + max(length))
genomicElementCols = c()
# genomicElementCols["homologousRegion"] = "#0AB45A"
# genomicElementCols["VariationWindows"] = "#FA7850"
# genomicElementCols["StableRegions"] = "black"
# genomicElementCols["HRPs"] = "#00A0FA"
#
# genomicElementCols["Telomere associated Repetitive Element 3"] = "#B07AA1"
# genomicElementCols["Telomere associated Repetitive Element 1"] = "#E15759"
genomicElementCols["TransitionPointWithChr5Dup"] = "#00E307"
genomicElementCols["DuplicatedRegion"] = "#2271B2"
genomicElementCols["VariationWindows"] = "#D55E00"
genomicElementCols["BiallelicSNPs"] = "#7CFFFA"
genomicElementCols["StableRegions"] = "black"
genomicElementCols["HRPs"] = "#3DB7E9"
genomicElementCols["Telomere associated Repetitive Element 3"] = "#F0E442"
genomicElementCols["Telomere associated Repetitive Element 1"] = "#F748A5"
combinedPlotColors = c(genomicElementCols,
descriptionColors)
chromPlot = ggplot() +
geom_rect(aes(xmin = minStart, xmax = newEnd,
ymin = 0, ymax = 1),
data = blank_chroms,
alpha = 0) +
geom_rect(
aes(
xmin = start,
xmax = end,
ymin = 1,
ymax = 1.25,
name = name,
length = length,
description = description,
ID = ID,
fill = description,
color = description
),
data = allGenes
) +
geom_rect(
aes(
xmin = minStart,
xmax = length,
ymin = 0,
ymax = 1,
),
fill = "grey60",
data = chroms_filt
) +
geom_rect(
aes(
xmin = X2,
xmax = X3,
ymin = 0,
ymax = 0.5,
name = name,
len = len,
start = X2,
end = X3,
color = "StableRegions",
fill = "StableRegions"
),
size = 0.011,
data = finalHRPII_HRPIII_windows_withTuned
) +
geom_rect(
aes(
xmin = X2,
xmax = X3,
ymin = 0.5,
ymax = 0.75,
name = name,
len = len,
start = X2,
end = X3,
color = "VariationWindows",
fill = "VariationWindows"
),
size = 0.01,
data = finalHRPII_HRPIII_windows_subWindows_withTuned
) +
geom_rect(
aes(
xmin = X2,
xmax = X3,
ymin = -0.25,
ymax = 0,
name = name,
len = len,
start = X2,
end = X3,
color = repeatType,
fill = repeatType
),
size = 0.01,
data = tare_filt %>%
mutate(len = X5, chrom = X1)
) +
geom_rect(
aes(
xmin = start,
xmax = end,
ymin = -0.25,
ymax = 0,
name = name,
len = len,
fill = "DuplicatedRegion",
color = "DuplicatedRegion"
),
data = shared_chrom11_chrom13
) +
geom_rect(
aes(
xmin = start,
xmax = end,
ymin = -0.25,
ymax = 0,
name = name,
len = len,
fill = "TransitionPointWithChr5Dup",
color = "TransitionPointWithChr5Dup",
start = start,
end = end
),
data = transitionPointWithChr5Dup
) +
geom_rect(
aes(
xmin = start,
xmax = end,
ymin = -0.25,
ymax = 0,
name = name,
length = length,
fill = "HRPs",
color = "HRPs"
),
data = allGenes_hrps
) +
geom_rect(
aes(
xmin = start,
xmax = end,
ymin = 0.75,
ymax = 1,
name = name,
AF = AF,
fill = "BiallelicSNPs",
color = "BiallelicSNPs"
),
# color = "black",
# fill = "black",
# color = "#D55E00",
# fill = "#D55E00",
data = variants_filt,
size = 0.01
) +
scale_fill_manual(values = combinedPlotColors) +
scale_color_manual(values = combinedPlotColors) +
facet_wrap(chrom ~ ., scale = "free", ncol = 1, strip.position = "left") +
labs(fill = "") +
sofonias_theme + theme(axis.text.y = element_blank(),
axis.ticks.y = element_blank())
# +
# scale_x_continuous(expand = c(0,0)) +
# scale_y_continuous(expand = c(0,0))The genomic locations for the windows investigated on chromosomes 8, 11, and 13
create_dt(finalHRPII_HRPIII_windows_withTuned_sum %>%
mutate(name = paste0(chrom, "-", minStart, "-", maxEnd),
length = maxEnd - minStart
))The genes in these regions
create_dt(allGenes)The telomere associated repetitive elements regions
create_dt(tare_filt)create_dt(
bind_rows(
transitionPointWithChr5Dup,
shared_chrom11_chrom13 %>%
mutate(name = "Duplicated region between chrom11 chrom13")
)
)chromPlotForFigure = chromPlot +
scale_fill_manual("Genes\nDescription",
values = combinedPlotColors,
breaks = names(descriptionColors),
labels = names(descriptionColors),
guide = guide_legend(
override.aes = list(color = descriptionColors,
fill = descriptionColors,
alpha = 1,
shape = 22,
size = 5),
nrow = 9,
order = 1)
) +
scale_color_manual("Genomic\nElements",
values = combinedPlotColors,
breaks = names(genomicElementCols),
labels = names(genomicElementCols),
guide = guide_legend(
override.aes = list(color = genomicElementCols,
fill = genomicElementCols,
alpha = 1,
shape = 22,
size = 5),
ncol = 2,
order = 2)
) +
theme(legend.text = element_text(size = 30), axis.text.x = element_text(size = 30, color = "black"),
plot.title = element_text(size = 30, color = "black"),
strip.text.y = element_text(size = 30, color = "black"),
legend.title = element_text(size = 30, face = "bold"),
legend.box="vertical", legend.margin=margin(),
legend.background = element_blank(),
legend.box.background = element_rect(colour = "black")) Orange blocks are regions within which there is variation and the black bars underneath them are relatively conserved regions between all strains. The dark blue bar shows where chromosome 11 and 13 have shared duplicated sequenced. The ends of the genomes without black bars represent non-core genome that are not preserved between different strains, the genes found within these regions are primarily steovrs, rifins, and PfEMP1s. Telomere associated repetitive elements (TARE) regions are shown as well.
print(chromPlotForFigure)
quartz_off_screen
2 Zoom in by click and dragging into the areas of interest, some of the variation windows are too small to see zoomed all the way out so you have to zoom in to see them.
ggplotly(chromPlot)chroms_filt_08_13 = chroms_filt %>%
filter(chrom %in% c("Pf3D7_08_v3", "Pf3D7_13_v3")) %>%
mutate(minStart = ifelse(chrom == "Pf3D7_08_v3", 1364235, minStart))%>%
mutate(minStart = ifelse(chrom == "Pf3D7_13_v3", 2830726, minStart))
blank_chroms_08_13 = blank_chroms %>%
filter(chrom %in% chroms_filt_08_13$chrom) %>%
mutate(minStart = ifelse(chrom == "Pf3D7_08_v3", 1364235, minStart))%>%
mutate(minStart = ifelse(chrom == "Pf3D7_13_v3", 2830726, minStart))
finalHRPII_HRPIII_windows_withTuned_08_13 = finalHRPII_HRPIII_windows_withTuned %>%
filter(chrom %in% chroms_filt_08_13$chrom) %>%
filter(!(X1 == "Pf3D7_08_v3" & X2 < 1364235))%>%
filter(!(X1 == "Pf3D7_13_v3" & X2 < 2830726))
tare_filt_08_13 = tare_filt %>%
filter(X1 %in% chroms_filt_08_13$chrom)
allGenes_08_13 = allGenes %>%
filter(chrom %in% chroms_filt_08_13$chrom)%>%
filter(!(chrom == "Pf3D7_08_v3" & start < 1364235))%>%
filter(!(chrom == "Pf3D7_13_v3" & start < 2830726))
chromPlot_08_13 = ggplot() +
geom_rect(aes(xmin = minStart, xmax = newEnd,
ymin = 0, ymax = 1),
data = blank_chroms_08_13,
alpha = 0) +
geom_rect(
aes(
xmin = start,
xmax = end,
ymin = 1,
ymax = 1.25,
name = name,
length = length,
description = description,
ID = ID,
fill = description,
color = description
),
data = allGenes_08_13
) +
geom_rect(
aes(
xmin = minStart,
xmax = length,
ymin = 0,
ymax = 1,
),
fill = "grey60",
data = chroms_filt_08_13
) +
geom_rect(
aes(
xmin = X2,
xmax = X3,
ymin = 0,
ymax = 1,
name = name,
len = len,
start = X2,
end = X3,
color = "StableRegions",
fill = "StableRegions"
),
size = 0.011,
data = finalHRPII_HRPIII_windows_withTuned_08_13
) +
geom_rect(
aes(
xmin = X2,
xmax = X3,
ymin = -0.25,
ymax = 0,
name = name,
len = len,
start = X2,
end = X3,
color = repeatType,
fill = repeatType
),
size = 0.011,
data = tare_filt_08_13 %>%
mutate(len = X5, chrom = X1)
)+
geom_rect(
aes(
xmin = start,
xmax = end,
ymin = -0.25,
ymax = 0,
name = name,
length = length,
fill = "HRPs",
color = "HRPs"
),
data = allGenes_hrps
) +
scale_fill_manual(values = combinedPlotColors) +
scale_color_manual(values = combinedPlotColors) +
facet_wrap(chrom ~ ., scale = "free", ncol = 1, strip.position = "left") +
labs(fill = "") +
sofonias_theme + theme(axis.text.y = element_blank(),
axis.ticks.y = element_blank()) +
scale_fill_manual("Genes\nDescription",
values = combinedPlotColors,
breaks = names(descriptionColors[names(descriptionColors) %in% allGenes_08_13$description]),
labels = names(descriptionColors[names(descriptionColors) %in% allGenes_08_13$description]),
guide = guide_legend(
override.aes = list(color = descriptionColors[names(descriptionColors) %in% allGenes_08_13$description],
fill = descriptionColors[names(descriptionColors) %in% allGenes_08_13$description],
alpha = 1,
shape = 22,
size = 5),
nrow = 5,
order = 1)
) +
scale_color_manual("Genomic\nElements",
values = combinedPlotColors,
breaks = names(genomicElementCols[c("StableRegions", "HRPs", "Telomere associated Repetitive Element 3", "Telomere associated Repetitive Element 1")]),
labels = names(genomicElementCols[c("StableRegions", "HRPs", "Telomere associated Repetitive Element 3", "Telomere associated Repetitive Element 1")]),
guide = guide_legend(
override.aes = list(color = genomicElementCols[c("StableRegions", "HRPs", "Telomere associated Repetitive Element 3", "Telomere associated Repetitive Element 1")],
fill = genomicElementCols[c("StableRegions", "HRPs", "Telomere associated Repetitive Element 3", "Telomere associated Repetitive Element 1")],
alpha = 1,
shape = 22,
size = 5),
ncol = 2,
order = 2)
) quartz_off_screen
2 One of the deletion patterns (13-5++) is associated with a transposed duplicated region of chr05 that includes pfmdr1 onto chromosome 13 which results in duplication of pfmdr1 and deletion of pfhrp3. These are the windows surrounding that duplication from chromosome 5.
pfmdr1_windows = readr::read_tsv("windows/stableWindows_05_regionOfInterestNarrow.bed", col_names = F) %>%
mutate(genomicID = paste0(X1, "-", X2, "-", X3)) %>%
mutate(chrom = X1, len = X3 -X2) %>%
rename(name = X4)
create_dt(pfmdr1_windows %>%
select(-chrom, -len) %>%
rename(chrom= X1, start = X2, end = X3, length = X5, strand = X6, geneInfo = X7))Local haplotype reconstruction was performed on each region for over 16,000 whole genome sequences (WGS) publicly available P. falciparum field samples. The called haplotypes were analyzed to determine which subregions contained variation in order to type each chromosome.
pfmdr1_windows_subWindows = readr::read_tsv("windows/stableWindows_05_regionOfInterestNarrow_allRefVariableRegions.bed", col_names = F) %>%
mutate(genomicID = paste0(X1, "-", X2, "-", X3)) %>%
mutate(chrom = X1, len = X3 - X2) %>%
rename(name = X4)
create_dt(pfmdr1_windows_subWindows %>%
select(-chrom, -len) %>%
rename(chrom= X1, start = X2, end = X3, length = X5, strand = X6, geneInfo = X7))chrom05_genes = readr::read_tsv("../MappingOutSurroundingRegions/surroundingRegionsMaterials/pfmdr1_region/regionBeds/split_Pf3D7_pfmdr1Region_genes.tab.txt", col_names = T)
allGenes_chrom05_genes = bind_rows(chrom05_genes) %>%
rename(chrom = col.0, start = col.1, end = col.2, name = col.3, length = col.4, strand = col.5) %>%
mutate(rawGeneDescription = description) %>%
mutate(gene = ifelse(grepl("PF3D7_0831800", description), "HRP II", "other")) %>%
mutate(gene = ifelse(grepl("PF3D7_1372200", description), "HRP III", gene)) %>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse("membrane associated erythrocyte binding-like protein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>%
mutate(description = ifelse("membrane associated erythrocyte binding-likeprotein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>%
mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description)) %>%
mutate(description = gsub(",putative", ", putative", description)) %>%
mutate(description = gsub(",pseudogene", ", pseudogene", description))%>%
mutate(description = gsub("unknownfunction", "unknown function", description))%>%
mutate(description = gsub("conserved protein, unknown function", "conserved Plasmodium protein, unknown function", description)) %>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description))%>%
mutate(description = gsub(",pseudogene", ", pseudogene", description))%>%
mutate(description = gsub("unknownfunction", "unknown function", description))%>%
mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>%
mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>%
mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>%
mutate(description = ifelse(grepl("sporozoite and liver stage tryptophan-rich protein, putative", description), "tryptophan/threonine-rich antigen", description))%>%
mutate(description = ifelse(grepl("CRA domain-containing protein, putative", description), "conserved Plasmodium protein, unknown function", description))%>%
mutate(description = gsub(",pseudogene", ", pseudogene", description)) %>%
mutate(description = gsub("surfaceantigen", "surface antigen", description)) %>%
mutate(description = gsub("Tetratricopeptide repeat, putative", "tetratricopeptide repeat protein, putative", description)) %>%
mutate(description = gsub("transmembraneprotein", "transmembrane protein", description)) %>%
mutate(description = ifelse(grepl("PfEMP1", description) & grepl("pseudogene", description), "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>%
mutate(description = gsub("PIR protein", "stevor", description)) %>%
mutate(description = gsub("erythrocyte membrane protein 1-like", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>%
mutate(description = gsub("acidic terminal segments, variant surface antigen of PfEMP1, putative", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description))%>%
mutate(description = ifelse(grepl("CoA binding protein", description, ignore.case = T), "acyl-CoA binding protein", description)) %>%
mutate(description = ifelse(grepl("transfer RNA", description) | grepl("tRNA", description), "tRNA", description))%>%
mutate(description = ifelse(grepl("cytoadherence", description), "CLAG", description))%>%
mutate(description = ifelse(grepl("surface-associated interspersed protein", description), "SURFIN", description))%>%
mutate(description = ifelse(grepl("SURFIN", description), "SURFIN", description))%>%
mutate(description = ifelse(grepl("stevor-like", description), "stevor, pseudogene", description)) %>%
mutate(description = ifelse(grepl("exported protein family", description), "exported protein family", description)) %>%
mutate(description = ifelse(grepl("ribosomal RNA", description), "rRNA", description)) %>%
mutate(description = ifelse(grepl("serine/threonine protein kinase", description), "serine/threonine protein kinase, FIKK family", description)) %>%
mutate(description = ifelse(grepl("hypothetical protein", description), "hypothetical protein, conserved", description)) %>%
mutate(description = ifelse(grepl("conserved Plasmodium protein, unknown function", description), "hypothetical protein, conserved", description)) %>%
mutate(description = ifelse(grepl("Rifin/stevor family, putative", description), "stevor", description)) %>%
mutate(description = ifelse(grepl("stevor", description), "stevor", description)) %>%
mutate(description = ifelse(grepl("rifin", description), "rifin", description)) %>%
mutate(description = ifelse(grepl("erythrocyte membrane protein 1 (PfEMP1), pseudogene", description), "erythrocyte membrane protein 1 (PfEMP1)", description)) %>%
mutate(description = ifelse(grepl("erythrocyte membrane protein 1", description), "erythrocyte membrane protein 1 (PfEMP1)", description)) %>%
mutate(description = ifelse(grepl("probably protein", description), "unspecified product", description)) %>%
mutate(description = ifelse(grepl("RESA", description), "RESA", description)) %>%
mutate(description = ifelse(grepl("ring-infected erythrocyte surface antigen", description), "ring-infected erythrocyte surface antigen", description))%>%
mutate(description = ifelse(grepl("Duffy binding domain/Erythrocyte binding antigen175, putative", description), "erythrocyte binding like protein 1", description)) %>%
mutate(description = ifelse(grepl("erythrocyte binding like protein 1", description), "erythrocyte binding like protein 1", description))%>%
mutate(description = ifelse(grepl("unspecified product", description), "hypothetical protein, conserved", description))%>%
mutate(description = ifelse(grepl("probable protein, unknown function", description), "hypothetical protein, conserved", description)) %>%
mutate(description = ifelse(grepl("RESA", description), "ring-infected erythrocyte surface antigen", description)) %>%
mutate(description = gsub("multidrug resistance protein 1", "multidrug resistance protein 1(pfmdr1)", description))
descriptionColorsNames = unique(c(allGenes_chrom05_genes$description))
descriptionColors = scheme$hex(length(descriptionColorsNames))
names(descriptionColors) = descriptionColorsNamespfmdr1_dupWithHrp3Del = tibble(
chrom = "Pf3D7_05_v3",
start = 952574,
end = 979203,
name = "pfmdr1_dup_with_hrp3_del",
len = 26629,
strand = '-'
)
pfmdr1_windows_sum = pfmdr1_windows %>%
group_by(chrom) %>%
summarise(minStart = min(X2))
pf3d7_region = pfmdr1_windows %>%
group_by(X1) %>%
summarise(X2 = min(X2),
X3 = max(X3)) %>%
mutate(name = paste0(X1, "-", X2, "-", X3),
length = X3 - X2,
strand = "+")
variants_pfmdr1_filt = gatk_calls_hrp2_3Regions_database_hrpsallMetaDeletionCalls %>%
filter(chrom %in% pfmdr1_windows$chrom) %>%
left_join(pfmdr1_windows_sum) %>%
filter(start >=minStart)
genomicElementCols = c()
# genomicElementCols["homologousRegion"] = "#0AB45A"
# genomicElementCols["VariationWindows"] = "#FA7850"
# genomicElementCols["StableRegions"] = "black"
# genomicElementCols["HRPs"] = "#00A0FA"
#
# genomicElementCols["Telomere associated Repetitive Element 3"] = "#B07AA1"
# genomicElementCols["Telomere associated Repetitive Element 1"] = "#E15759"
genomicElementCols["Chr5Dup"] = "#EF0096"
genomicElementCols["VariationWindows"] = "#D55E00"
genomicElementCols["BiallelicSNPs"] = "#7CFFFA"
genomicElementCols["StableRegions"] = "black"
# genomicElementCols["HRPs"] = "#3DB7E9"
#
# genomicElementCols["Telomere associated Repetitive Element 3"] = "#F0E442"
# genomicElementCols["Telomere associated Repetitive Element 1"] = "#F748A5"
combinedPlotColors = c(genomicElementCols,
descriptionColors)
chromPlot_chr05 = ggplot() +
geom_rect(
aes(
xmin = start,
xmax = end,
ymin = 1,
ymax = 1.25,
name = name,
length = length,
description = description,
ID = ID,
fill = description,
color = description
),
data = allGenes_chrom05_genes
) +
geom_rect(
aes(
xmin = X2,
xmax = X3,
ymin = 0,
ymax = 1,
),
fill = "grey60",
data = pf3d7_region
) +
geom_rect(
aes(
xmin = X2,
xmax = X3,
ymin = 0,
ymax = 0.5,
name = name,
len = len,
start = X2,
end = X3,
color = "StableRegions",
fill = "StableRegions"
),
size = 0.011,
data = pfmdr1_windows
) +
geom_rect(
aes(
xmin = X2,
xmax = X3,
ymin = 0.5,
ymax = 0.75,
name = name,
len = len,
start = X2,
end = X3,
color = "VariationWindows",
fill = "VariationWindows"
),
size = 0.01,
data = pfmdr1_windows_subWindows
)+
geom_rect(
aes(
xmin = start,
xmax = end,
ymin = -0.25,
ymax = 0,
name = name,
len = len,
fill = "Chr5Dup",
color = "Chr5Dup"
),
data = pfmdr1_dupWithHrp3Del
) +
geom_rect(
aes(
xmin = start,
xmax = end,
ymin = 0.75,
ymax = 1,
name = name,
AF = AF,
fill = "BiallelicSNPs",
color = "BiallelicSNPs"
),
# color = "black",
# fill = "black",
# color = "#D55E00",
# fill = "#D55E00",
data = variants_pfmdr1_filt,
size = 0.01
) +
scale_fill_manual(values = combinedPlotColors) +
scale_color_manual(values = combinedPlotColors) +
facet_wrap(chrom ~ ., scale = "free", ncol = 1, strip.position = "left") +
labs(fill = "") +
sofonias_theme + theme(axis.text.y = element_blank(),
axis.ticks.y = element_blank())The Pf3D7 region around pfmdr1 that was investigated and the region that is duplicated
create_dt(bind_rows(pf3d7_region %>%
rename(chrom = X1,
start = X2,
end = X3,
len = length)
))create_dt(allGenes_chrom05_genes)create_dt(pfmdr1_dupWithHrp3Del)chromPlot_chr05ForFigure = chromPlot_chr05 +
scale_fill_manual("Genes\nDescription",
values = combinedPlotColors,
breaks = names(descriptionColors),
labels = names(descriptionColors),
guide = guide_legend(
override.aes = list(color = descriptionColors,
fill = descriptionColors,
alpha = 1,
shape = 22,
size = 5),
nrow = 7,
order = 1)
) +
scale_color_manual("Genomic\nElements",
values = combinedPlotColors,
breaks = names(genomicElementCols),
labels = names(genomicElementCols),
guide = guide_legend(
override.aes = list(color = genomicElementCols,
fill = genomicElementCols,
alpha = 1,
shape = 22,
size = 5),
ncol = 2,
order = 2)
) +
theme(legend.text = element_text(size = 30), axis.text.x = element_text(size = 30, color = "black"),
plot.title = element_text(size = 30, color = "black"),
strip.text.y = element_text(size = 30, color = "black"),
legend.title = element_text(size = 30, face = "bold"),
legend.box="vertical", legend.margin=margin(),
legend.background = element_blank(),
legend.box.background = element_rect(colour = "black")) Orange blocks are regions within which there is variation and the black bars underneath them are relatively conserved regions between all strains.
print(chromPlot_chr05ForFigure)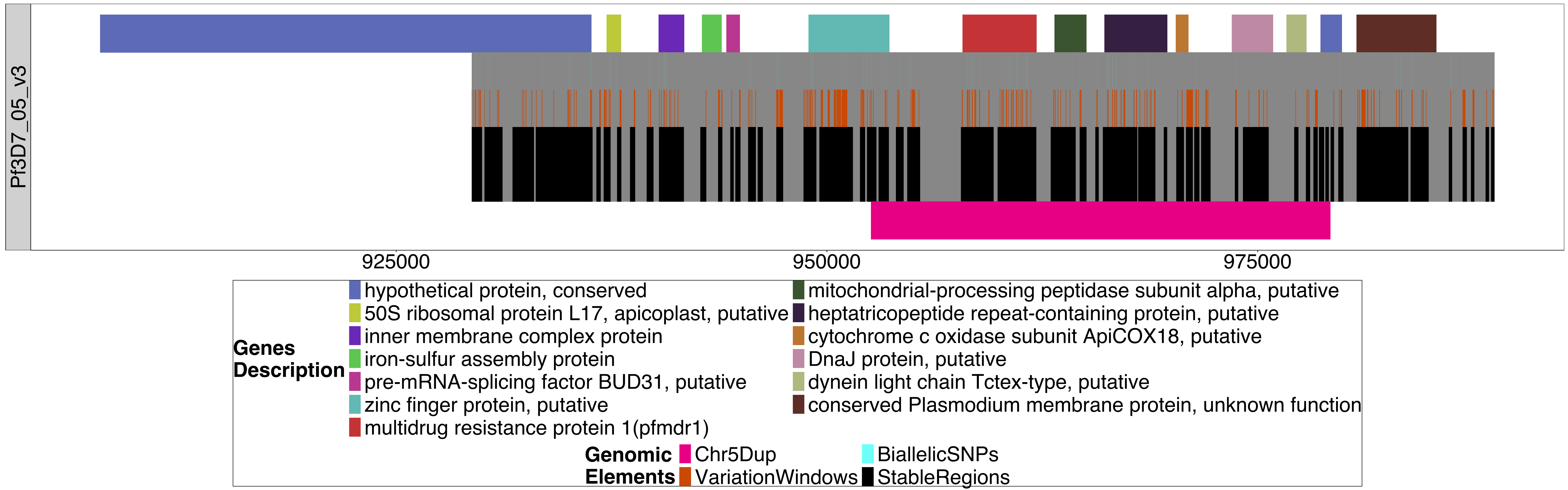
quartz_off_screen
2 Zoom in by click and dragging into the areas of interest, some of the variation windows are too small to see zoomed all the way out so you have to zoom in to see them.
ggplotly(chromPlot_chr05)combined_large_windows = bind_rows(pfmdr1_windows, finalHRPII_HRPIII_windows_withTuned) %>%
select(-genomicID, -chrom, -len)
colnames(combined_large_windows) = c(
"Chromosome",
"Start Position",
"End Position",
"Chromosome: Start-End Position",
"Window Length",
"Strand (+/-)",
"Functional Annotation"
)
write_tsv(combined_large_windows, "Table S2 Non-Paralogous windows.tsv")
combined_large_windows_subWindows = bind_rows(pfmdr1_windows_subWindows,
finalHRPII_HRPIII_windows_subWindows_withTuned) %>%
select(-genomicID, -chrom, -len)
colnames(combined_large_windows_subWindows) = c(
"Chromosome",
"Start Position",
"End Position",
"Chromosome: Start-End Position",
"Window Length",
"Strand (+/-)",
"Functional Annotation"
)
write_tsv(combined_large_windows_subWindows,
"Table S3 Variations in non-paralog windows.tsv")
combined_variantsOut = variants_pfmdr1_filt %>%
select(1:4, ref, alts) %>%
bind_rows(variants_filt %>%
select(1:4, ref, alts))
colnames(combined_variantsOut)[1:4] = c(
"Chromosome",
"Start Position",
"End Position",
"Chromosome: Start-End Position")
write_tsv(combined_variantsOut,
"Table S4 Biallelic SNPs.tsv")