meta.tab.txtmetaByBioSamplereal_mccoil_COI_calls.tsvallBasicInfo_filt_sp_mat.RdatarowAnnoColors.RdataallCov_summaryStats.tab.txt.gz
Plotting coverage in sub windows pfhrp3 deletion
Setup
Downloads
Code
meta = readr::read_tsv("../meta/metadata/meta.tab.txt") %>%
mutate(country = gsub("South East Asia - East", "Cambodia", country))
metaByBioSample = readr::read_tsv("../meta/metadata/metaByBioSample.tab.txt") %>%
mutate(country = gsub("South East Asia - East", "Cambodia", country))
realmccoilCoiCalls = readr::read_tsv("wgs_variants/THEREALMcCOIL/categorical_method/real_mccoil_COI_calls.tsv")
realmccoilCoiCalls_poly = realmccoilCoiCalls %>%
filter(random_median != 1 | topHE_median != 1)
allMetaDeletionCalls = readr::read_tsv("allMetaDeletionCalls.tab.txt") %>%
filter(BiologicalSample %!in% realmccoilCoiCalls_poly$BiologicalSample)
hrp3Pat2_samplesWithTare1 = readr::read_tsv("deletion_patterns/hrp3Pat2_samplesWithTare1.tsv")
# char11 tare1
chr11_tare_calls = readr::read_tsv("deletion_patterns/finalHRPII_HRPIII_windows_investTelemeraseHealing_chr11_full_out_step50_windowSize100_rowAnno.tsv")
chr11_tare_calls_hasTare1 = chr11_tare_calls %>%
filter(`TARE1 On Chr11`)
# these are only pattern 2 aka chr11/chr13-
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_sampleRegion_hrp3_pat2_sample_deletion_meta = readr::read_tsv("deletion_patterns/stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_sampleRegion_hrp3_pat2_sample_deletion_meta.tsv")
stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_sampleRegion_hrp3_pat2_sample_deletion_meta = stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_sampleRegion_hrp3_pat2_sample_deletion_meta %>%
mutate(Pattern = case_when(
`TARE1 On Chr5` | `Transposition With Chr5` ~ "13-5++",
`TARE1 On Chr13` ~ "13-TARE1",
T ~ NA
))
# mutate(Pattern = case_when(
# `TARE1 On Chr5` | `Transposition With Chr5` ~ "5++/13-",
# `TARE1 On Chr13` ~ "11/13-TARE1",
# T ~ NA
# ))
allMetaDeletionCalls_chr11_deleted = allMetaDeletionCalls %>%
filter(possiblyChr11Deleted)%>%
left_join(stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_sampleRegion_hrp3_pat2_sample_deletion_meta %>%
select(sample, Pattern)) %>%
mutate(Pattern = ifelse(HRP3_deletionPattern == "Pattern 1", "13-11++", Pattern))%>%
mutate(Pattern = ifelse(is.na(Pattern) & sample %in% chr11_tare_calls_hasTare1$sample, "11-TARE1", Pattern))
# mutate(Pattern = ifelse(HRP3_deletionPattern == "Pattern 1", "11++/13-", Pattern))%>%
# mutate(Pattern = ifelse(is.na(Pattern) & sample %in% chr11_tare_calls_hasTare1$sample, "11-TARE1/13", Pattern))
allMetaDeletionCalls_hrp2_deleted = allMetaDeletionCalls %>%
filter(possiblyHRP2Deleted)%>%
left_join(stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_sampleRegion_hrp3_pat2_sample_deletion_meta %>%
select(sample, Pattern)) %>%
mutate(Pattern = ifelse(HRP3_deletionPattern == "Pattern 1", "13-11++", Pattern))%>%
mutate(Pattern = ifelse(is.na(Pattern) & sample %in% chr11_tare_calls_hasTare1$sample, "11-TARE1", Pattern))
# mutate(Pattern = ifelse(HRP3_deletionPattern == "Pattern 1", "11++/13-", Pattern))%>%
# mutate(Pattern = ifelse(is.na(Pattern) & sample %in% chr11_tare_calls_hasTare1$sample, "11-TARE1/13", Pattern))
allMetaDeletionCalls_hrp3_deleted = allMetaDeletionCalls %>%
filter(possiblyHRP3Deleted)%>%
left_join(stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200_sampleRegion_hrp3_pat2_sample_deletion_meta %>%
select(sample, Pattern)) %>%
mutate(Pattern = ifelse(HRP3_deletionPattern == "Pattern 1", "13-11++", Pattern))%>%
mutate(Pattern = ifelse(is.na(Pattern) & sample %in% chr11_tare_calls_hasTare1$sample, "11-TARE1", Pattern)) %>%
# mutate(Pattern = ifelse(HRP3_deletionPattern == "Pattern 1", "11++/13-", Pattern)) %>%
# mutate(Pattern = ifelse(is.na(Pattern) & sample %in% chr11_tare_calls_hasTare1$sample, "11-TARE1/13", Pattern)) %>%
mutate(Pattern = ifelse(is.na(Pattern), "13-", Pattern))
#mutate(Pattern = factor(Pattern, levels = c("13-11++", "13-5++", "13-TARE1", "13-"))) %>%Sample Meta data
Code
cov = readr::read_tsv("../meta/allCov_summaryStats.tab.txt.gz")
meta = readr::read_tsv("../meta/metadata/meta.tab.txt")
masterTable = readr::read_tsv("../meta/metadata/masterTable.tab.txt")
coveredSamples = readr::read_tsv("samplesCovered.txt", col_names = "sample") %>%
left_join(meta %>%
select(sample, ExperimentSample, BiologicalSample))
# chr07_dup_samplesCovered = readr::read_tsv("rRNA_chr05_chr07/samplesCovered.txt", col_names = c("sample"))
# chr07_dup = readr::read_tsv("rRNA_chr05_chr07/possible_chrom5_deletion_samples.txt", col_names = c("sample")) %>%
# left_join(cov %>%
# select(sample, meanPerBaseCov)) %>%
# left_join(masterTable)%>%
# mutate(chr05chr07rRNAPattern = "05-/07++")
deletionCalls = allMetaDeletionCalls %>%
select(-HRP3_deletionPattern) %>%
left_join(allMetaDeletionCalls_hrp3_deleted %>%
ungroup() %>%
select(sample, Pattern)) %>%
mutate(Pattern = ifelse(is.na(Pattern) & sample %in% chr11_tare_calls_hasTare1$sample, "11-TARE1", Pattern)) %>%
# mutate(Pattern = ifelse(is.na(Pattern) & sample %in% chr11_tare_calls_hasTare1$sample, "11-TARE1/13", Pattern)) %>%
left_join(cov %>%
select(sample, meanPerBaseCov))
# %>%
# full_join(chr07_dup)
meta_allOthers = meta %>%
filter(BiologicalSample %!in% deletionCalls$BiologicalSample)%>%
left_join(cov %>%
select(sample, meanPerBaseCov)) %>%
left_join(masterTable %>%
select(sample, SRARuns))%>%
filter(PreferredSample) %>%
select(-sample, -ExperimentSample,-PreferredSample) %>%
rename(sample = BiologicalSample) %>%
filter(!is.na(SRARuns))
meta_withDeletionMetaOutput = bind_rows(
deletionCalls %>%
select(-sample, -ExperimentSample,-PreferredSample) %>%
rename(sample = BiologicalSample) ,
meta_allOthers
) %>%
mutate(uncallable = sample %!in% coveredSamples$BiologicalSample) %>%
rename(HRP2Call = possiblyHRP2Deleted,
HRP3Call = possiblyHRP3Deleted,
HRPsCall = hrpCall,
Chr11TelemericCall = possiblyChr11Deleted) %>%
mutate(HRP2Call = case_when(uncallable ~ "uncallable",
is.na(HRP2Call) ~ "present",
!is.na(HRP2Call) & HRP2Call ~ "deletion",
T ~ "present"),
HRP3Call = case_when(uncallable ~ "uncallable",
is.na(HRP3Call) ~ "present",
!is.na(HRP3Call) & HRP3Call ~ "deletion",
T ~ "present"),
HRPsCall = case_when(uncallable ~ "uncallable",
is.na(HRPsCall) ~ "pfhrp2+/pfhrp3+",
T ~ HRPsCall),
Chr11TelemericCall = case_when(uncallable ~ "uncallable",
Chr11TelemericCall ~ "deletion",
is.na(Chr11TelemericCall) ~ "present",
T ~ "uncallable"
)
)
# %>%
# mutate(chr05chr07rRNAPattern = case_when(sample %in% chr07_dup$sample ~ "05-/07++",
# sample %in% chr07_dup_samplesCovered$sample ~ "05/07",
# T ~ "uncallable"))
masterTable_withDeletionMetaOutput_fieldSamples_labIsolates = meta_withDeletionMetaOutput %>% filter(IsFieldSample | site == "LabIsolate")
create_dt(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates)Code
write_tsv(meta_withDeletionMetaOutput, "sample_metadata_withAllDeletionCalls.tsv")
write_tsv(meta_withDeletionMetaOutput, "Supplemental Table - sample metadata.tsv")
write_tsv(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates, "Supplemental Table - sample metadata - field samples and Lab Isolates.tsv")Deletion Counts
Continent
HRP2
Code
masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp2 = masterTable_withDeletionMetaOutput_fieldSamples_labIsolates %>%
filter(HRP2Call == "deletion") %>%
group_by(secondaryRegion) %>%
rename(continent = secondaryRegion) %>%
count() %>%
group_by() %>%
mutate(total = sum(n))%>%
mutate(percentage = n * 100/total)
write_tsv(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp2, "hrp2_continent_deletion_counts.tsv")
create_dt(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp2)HRP3
Code
masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp3 = masterTable_withDeletionMetaOutput_fieldSamples_labIsolates %>%
filter(HRP3Call == "deletion") %>%
group_by(secondaryRegion) %>%
rename(continent = secondaryRegion) %>%
count() %>%
group_by() %>%
mutate(total = sum(n)) %>%
mutate(percentage = n * 100/total)
write_tsv(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp3, "hrp3_continent_deletion_counts.tsv")
create_dt(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp3)HRP3 Pattern
Code
masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp3_pattern = masterTable_withDeletionMetaOutput_fieldSamples_labIsolates %>%
filter(HRP3Call == "deletion") %>%
group_by(secondaryRegion, Pattern) %>%
rename(continent = secondaryRegion) %>%
count() %>%
group_by(Pattern) %>%
mutate(PatternTotal = sum(n, na.rm = T)) %>%
mutate(PatternPercentage = n * 100/PatternTotal) %>%
group_by(continent) %>%
mutate(continentTotal = sum(n)) %>%
mutate(continentPercentage = n * 100/continentTotal) %>%
group_by() %>%
mutate(total = sum(n)) %>%
mutate(percentage = n * 100/total)
write_tsv(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp3_pattern, "hrp3_pattern_continent_deletion_counts.tsv")
create_dt(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp3_pattern)Code
masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp3_pattern_raw = masterTable_withDeletionMetaOutput_fieldSamples_labIsolates %>%
filter(HRP3Call == "deletion") %>%
group_by(Pattern) %>%
count() %>%
group_by() %>%
mutate(PatternTotal = sum(n, na.rm = T)) %>%
mutate(percentage = n * 100/PatternTotal)
write_tsv(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp3_pattern_raw, "hrp3_pattern_deletion_counts.tsv")
create_dt(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp3_pattern_raw)HRP2 and HRP3
Code
masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp2_and_hrp3 = masterTable_withDeletionMetaOutput_fieldSamples_labIsolates %>%
filter(HRP2Call == "deletion" & HRP3Call == "deletion") %>%
group_by(secondaryRegion, Pattern) %>%
rename(continent = secondaryRegion) %>%
count() %>%
group_by() %>%
mutate(total = sum(n)) %>%
mutate(percentage = n * 100/total)
write_tsv(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp2_and_hrp3, "hrp2_and_hrp3_continent_deletion_counts.tsv")
create_dt(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrp2_and_hrp3)Chr11 Deletion
Code
masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_chr11 = masterTable_withDeletionMetaOutput_fieldSamples_labIsolates %>%
filter(Chr11TelemericCall == "deletion") %>%
group_by(secondaryRegion) %>%
rename(continent = secondaryRegion) %>%
count() %>%
group_by() %>%
mutate(total = sum(n)) %>%
mutate(percentage = n * 100 / total)
write_tsv(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_chr11, "chr11_continent_deletion_counts.tsv")
create_dt(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_chr11)HRP Calls
Code
masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrpCalls = masterTable_withDeletionMetaOutput_fieldSamples_labIsolates %>%
group_by(HRPsCall) %>%
count() %>%
group_by() %>%
mutate(total = sum(n)) %>%
mutate(percentage = n * 100 / total)
write_tsv(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrpCalls, "hrpCalls_deletion_counts.tsv")
create_dt(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_hrpCalls)Pattern
Code
masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_pattern = masterTable_withDeletionMetaOutput_fieldSamples_labIsolates %>%
group_by(Pattern) %>%
count() %>%
group_by() %>%
mutate(total = sum(n)) %>%
mutate(percentage = n * 100 / total)
write_tsv(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_pattern, "pattern_counts.tsv")
create_dt(masterTable_withDeletionMetaOutput_fieldSamples_labIsolates_pattern)Plotting Coverage
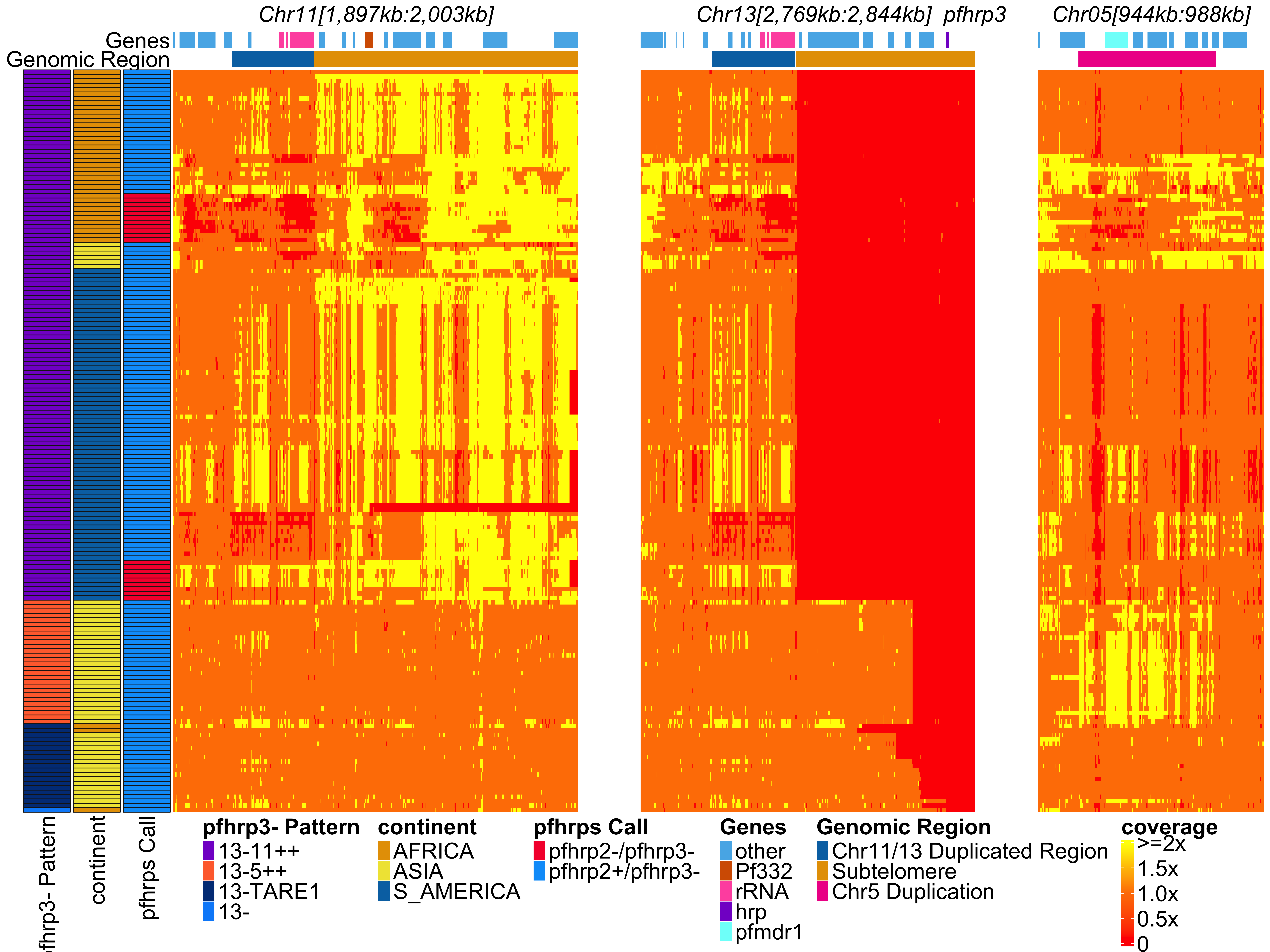
Coverage of genomic regions invovled in pfhrp3 deletions of samples with pfhrp3 deletions
Displaying from Pf3D7 genomic version=2015-06-18 of the following chromosome regions
- chromosome 05: 944389 - 988747 944kb:988kb
- chromosome 11: 1897157 - 2003328 1,897kb:2,003kb
- chromosome 13: 2769916 - 2844777 2,769kb:2,844kb
Code
allBasicInfo_filt_sp_mat_hrp3_deleted = allBasicInfo_filt_sp_mat[rownames(allBasicInfo_filt_sp_mat) %in% allMetaDeletionCalls_hrp3_deleted$sample, ]
metaSelected_hrp3_deleted = allMetaDeletionCalls_hrp3_deleted[match(rownames(allBasicInfo_filt_sp_mat_hrp3_deleted), allMetaDeletionCalls_hrp3_deleted$sample), ]
allBasicInfo_filt_sp_mat_hrp3_deleted_hclust = hclust(dist(allBasicInfo_filt_sp_mat_hrp3_deleted))
allBasicInfo_filt_sp_mat_hrp3_deleted_hclust_ordering = tibble(
sample = rownames(allBasicInfo_filt_sp_mat_hrp3_deleted)[allBasicInfo_filt_sp_mat_hrp3_deleted_hclust$order],
hclustInputOrder = allBasicInfo_filt_sp_mat_hrp3_deleted_hclust$order,
hclustOutputOrder = 1:nrow(allBasicInfo_filt_sp_mat_hrp3_deleted)
)
rowAnnoDf_hrp3_deleted = metaSelected_hrp3_deleted %>%
left_join(allBasicInfo_filt_sp_mat_hrp3_deleted_hclust_ordering) %>%
select("Pattern", "secondaryRegion", "hrpCall", hclustOutputOrder) %>%
rename(continent = secondaryRegion) %>%
as.data.frame()
rowAnnoColorsMod = rowAnnoColors
rowAnnoColorsMod[["Pattern"]] = rowAnnoColorsMod_hrp3DeletionPattern
rowAnnoColorsMod[["pfhrp3- Pattern"]] = rowAnnoColorsMod_hrp3DeletionPattern
rowAnnoColorsMod[["DeletionPattern"]] = rowAnnoColorsMod_hrp3DeletionPattern
rowAnnoColorsMod[["pfhrps Call"]] = pfhrpsCallColors
topAnnoColors_mod = list()
topAnnoColors_mod[["Genes"]] = c("other" = "#56B4E9", hrp = "#009E73", Pf332 = "#D55E00", rRNA = "#FF5AAF", "pfmdr1" = "#7CFFFA")
topAnnoColors_mod[["Genes"]] = c("other" = "#56B4E9", hrp = "#8400CD", Pf332 = "#D55E00", rRNA = "#FF5AAF", "pfmdr1" = "#7CFFFA")
topAnnoColors_mod[["Genomic Region"]] = c("Chr11/13 Duplicated Region" = "#0072B2", "Subtelomere" = "#E69F00", "Chr5 Duplication" = "#EF0096")
rowAnnoDf_hrp3_deleted_sorted = rowAnnoDf_hrp3_deleted %>%
as_tibble() %>%
mutate(rowID = row_number()) %>%
#mutate(Pattern = factor(Pattern, levels = c("11++/13-", "5++/13-", "11/13-TARE1", "13-"))) %>%
#mutate(Pattern = gsub("11/13-TARE1", "13-")) %>%
mutate(Pattern = factor(Pattern, levels = c("13-11++", "13-5++", "13-TARE1", "13-"))) %>%
arrange(Pattern, continent, hrpCall, hclustOutputOrder) %>%
rename("pfhrps Call" = hrpCall)
rowAnnoDf_hrp3_deleted_sortedDf = rowAnnoDf_hrp3_deleted_sorted %>%
select(-rowID, -hclustOutputOrder) %>%
as.data.frame()
allBasicInfo_filt_sp_mat_noLabs = allBasicInfo_filt_sp_mat_hrp3_deleted
rownames(allBasicInfo_filt_sp_mat_noLabs) = NULL
colnames(allBasicInfo_filt_sp_mat_noLabs) = NULL
allBasicInfo_filt_sp_mat_noLabs_chr08 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_08_", colnames(allBasicInfo_filt_sp_mat))] #08: 1290365 - 1387982 1,290kb:1,387kb
allBasicInfo_filt_sp_mat_noLabs_chr11 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_11_", colnames(allBasicInfo_filt_sp_mat))] #11: 1897157 - 2003328 1,897kb:2,003kb
allBasicInfo_filt_sp_mat_noLabs_chr13 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_13_", colnames(allBasicInfo_filt_sp_mat))] #13: 2769916 - 2844777 2,769kb:2,844kb
#05: 944389 - 988747 944kb:988kb
#08: 1290365 - 1387982 1,290kb:1,387kb
#11: 1897157 - 2003328 1,897kb:2,003kb
#13: 2769916 - 2844777 2,769kb:2,844kb
allBasicInfo_filt_sp_mat_noLabs_chr11_sorted = allBasicInfo_filt_sp_mat_noLabs_chr11[rowAnnoDf_hrp3_deleted_sorted$rowID,]
allBasicInfo_filt_sp_mat_noLabs_chr13_sorted = allBasicInfo_filt_sp_mat_noLabs_chr13[rowAnnoDf_hrp3_deleted_sorted$rowID,]
rowAnnoDf_hrp3_deleted_sortedDf_mod = rowAnnoDf_hrp3_deleted_sortedDf
rowAnnoDf_hrp3_deleted_sortedDf_mod = rowAnnoDf_hrp3_deleted_sortedDf_mod%>%
rename("pfhrp3- Pattern" = "Pattern")
annotationTextSize = 20
sideAnno = rowAnnotation(
df = rowAnnoDf_hrp3_deleted_sortedDf_mod,
col = rowAnnoColorsMod,
gp = gpar(col = "grey10"),
simple_anno_size = unit(1.5, "cm"),
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
na_col = c("#99999900"),
gap = unit(0.1, "cm")
)
topAnnoDf = regions[,c("inGene", "genomicRegion", "chrom")] %>% as.data.frame()
topAnnoDf_chr08 = topAnnoDf %>% filter(grepl("_08_",chrom)) %>% select(-chrom)
topAnnoDf_chr11 = topAnnoDf %>% filter(grepl("_11_",chrom)) %>% select(-chrom)
topAnnoDf_chr13 = topAnnoDf %>% filter(grepl("_13_",chrom)) %>% select(-chrom)
topAnnoDf_chr11_mod = topAnnoDf_chr11 %>%
rename("Genomic Region" = genomicRegion, "Genes" = "inGene") %>%
mutate(Genes = ifelse(Genes, "other", NA)) %>%
mutate(Genes = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "rRNA"), `Genomic Region`, Genes)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "After Duplicated Region"), "Subtelomere", `Genomic Region`))%>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("rRNA", "Duplicated Region"), "Chr11/13 Duplicated Region", `Genomic Region`))%>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("other"), NA, `Genomic Region`))
topAnnoDf_chr13_mod = topAnnoDf_chr13 %>%
rename("Genomic Region" = genomicRegion, "Genes" = "inGene")%>%
mutate(Genes = ifelse(Genes, "other", NA)) %>%
mutate(Genes = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "rRNA"), `Genomic Region`, Genes)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "After Duplicated Region"), "Subtelomere", `Genomic Region`))%>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("rRNA", "Duplicated Region"), "Chr11/13 Duplicated Region", `Genomic Region`)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("other"), NA, `Genomic Region`))
topAnno_chr11 = HeatmapAnnotation(
df = topAnnoDf_chr11_mod,
col = topAnnoColors_mod,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
show_annotation_name = T,
na_col = "#FFFFFF00",
gap = unit(0.1, "cm")
)
topAnno_chr13 = HeatmapAnnotation(
df = topAnnoDf_chr13_mod,
col = topAnnoColors_mod,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
show_annotation_name = F,
na_col = "#FFFFFF00",
gap = unit(0.1, "cm")
)
library(circlize)
col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3)))
allCoverageHeatMap_chr11 = Heatmap(
allBasicInfo_filt_sp_mat_noLabs_chr11_sorted,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno_chr11,
left_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
title = "coverage", at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
column_title = "Chr11[1,897kb:2,003kb]",
column_title_gp = gpar(fontsize = 20, fontface = "italic")
)
allCoverageHeatMap_chr13 = Heatmap(
allBasicInfo_filt_sp_mat_noLabs_chr13_sorted,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno_chr13,
# left_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold",
title = "coverage", at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x"))
),
column_title = " Chr13[2,769kb:2,844kb] pfhrp3",
column_title_gp = gpar(fontsize = 20, fontface = "italic")
)Reading in chromosome 5 pfmdr1 dup region
Code
cov = readr::read_tsv("../meta/allCov_summaryStats.tab.txt.gz")
allReports_chr5 = readr::read_tsv("deletion_patterns/stableWindows_05_regionOfInterestNarrow_full_step150_windowSize200/popClustering/reports/allBasicInfo.tab.txt.gz") %>%
filter(start >= 944389) %>%
#filter(sample %in% allMetaDeletionCalls_Hrp3pattern2_samples$sample) %>%
filter(sample %in% rownames(allBasicInfo_filt_sp_mat_hrp3_deleted)) %>%
mutate(inGene = !is.na(extraField0) ) %>%
left_join(cov) %>%
mutate(medianCov = median(perBaseCoverage),
meanCov = mean(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = ifelse(inGene, perBaseCoverage/medianPerBaseCov_inGenes, perBaseCoverage/medianPerBaseCov_notInGenes)) %>%
mutate(perBaseCoverageNormRounded = ifelse(perBaseCoverageNorm >0.10 & perBaseCoverageNorm <= 1, 1, perBaseCoverageNorm)) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNormRounded))
allReports_chr5_sp = allReports_chr5 %>%
select(sample, name, perBaseCoverageNormRounded) %>%
spread(name, perBaseCoverageNormRounded)
allReports_chr5_sp_mat = as.matrix(allReports_chr5_sp[,2:ncol(allReports_chr5_sp)])
rownames(allReports_chr5_sp_mat) = allReports_chr5_sp$sample
allReports_chr5_sp_mat = allReports_chr5_sp_mat[match(rownames(allBasicInfo_filt_sp_mat_hrp3_deleted)[rowAnnoDf_hrp3_deleted_sorted$rowID], rownames(allReports_chr5_sp_mat)), ]
allReports_chr5_sp_mat[allReports_chr5_sp_mat >2] = 2
topAnno_chr05 = allReports_chr5 %>%
filter(sample == .$sample[1]) %>%
select(name, extraField0) %>%
rename(`Gene Description` = extraField0) %>%
mutate(`Gene Description` = gsub(".*description=", "", `Gene Description`))%>%
mutate(`Gene Description` = gsub("\\].*", "", `Gene Description`)) %>%
mutate(`Gene Description` = ifelse(grepl("PHIST", `Gene Description`), gsub("\\).*", "", gsub(".*PHIST", "PHIST", `Gene Description`)), `Gene Description`))
topAnno_chr05Df = topAnno_chr05 %>%
separate(name, into = c("chrom", "start", "end"), remove = F, convert = T, sep = "-") %>%
mutate(`Genomic Region` = ifelse(start >= 952474 & end <= 979431, "Chr5 Duplication", NA)) %>%
select(-name, -chrom, -start, -end) %>%
as.data.frame()
topAnno_chr05Colors = createColorListFromDf(topAnno_chr05Df)
# 05: 944389 - 988747 944kb:988kb
topAnno_chr05Df_mod = topAnno_chr05Df %>%
select(`Gene Description`, `Genomic Region`) %>%
mutate(Genes = ifelse(is.na(`Gene Description`), NA, "other")) %>%
mutate(Genes = ifelse(`Gene Description` == "multidrug resistance protein 1", "pfmdr1", Genes)) %>%
select(`Genes`, `Genomic Region`)
topAnno_chr05HM = HeatmapAnnotation(
df = topAnno_chr05Df_mod,
col = topAnnoColors_mod,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
na_col = c("#99999900"),
gap = unit(0.1, "cm"),
show_annotation_name = F
)
allReports_chr5_sp_mat_nolab = allReports_chr5_sp_mat
colnames(allReports_chr5_sp_mat_nolab) = NULL
rownames(allReports_chr5_sp_mat_nolab) = NULL
allReports_chr5_sp_mat_hm = Heatmap(
allReports_chr5_sp_mat_nolab,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno_chr05HM,
#right_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold",
title = "coverage", at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x"))
),
column_title = "Chr05[944kb:988kb]",
column_title_gp = gpar(fontsize = 20, fontface = "italic")
)Code
#05: 944389 - 988747 944kb:988kb
#11: 1897157 - 2003328 1,897kb:2,003kb
#13: 2769916 - 2844777 2,769kb:2,844kb
hms_list = allCoverageHeatMap_chr11 + allCoverageHeatMap_chr13 + allReports_chr5_sp_mat_hm
draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom",
ht_gap = unit(2, "cm"),
#padding = unit(c(5.5 *5*2, 5.5, 5.5, 5.5), "points")
)
Code
#05: 944389 - 988747 944kb:988kb
#11: 1897157 - 2003328 1,897kb:2,003kb
#13: 2769916 - 2844777 2,769kb:2,844kb
hms_list = allCoverageHeatMap_chr11 + allCoverageHeatMap_chr13 + allReports_chr5_sp_mat_hm
pdf("allHeatmap_byChrom_filtered_chr11_chr13_chr5_sorted.pdf", useDingbats = F, width = 16, height = 12)
draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom",
ht_gap = unit(2, "cm"),
padding = unit(c(5.5 *5, 5.5, 5.5, 5.5), "points")
)
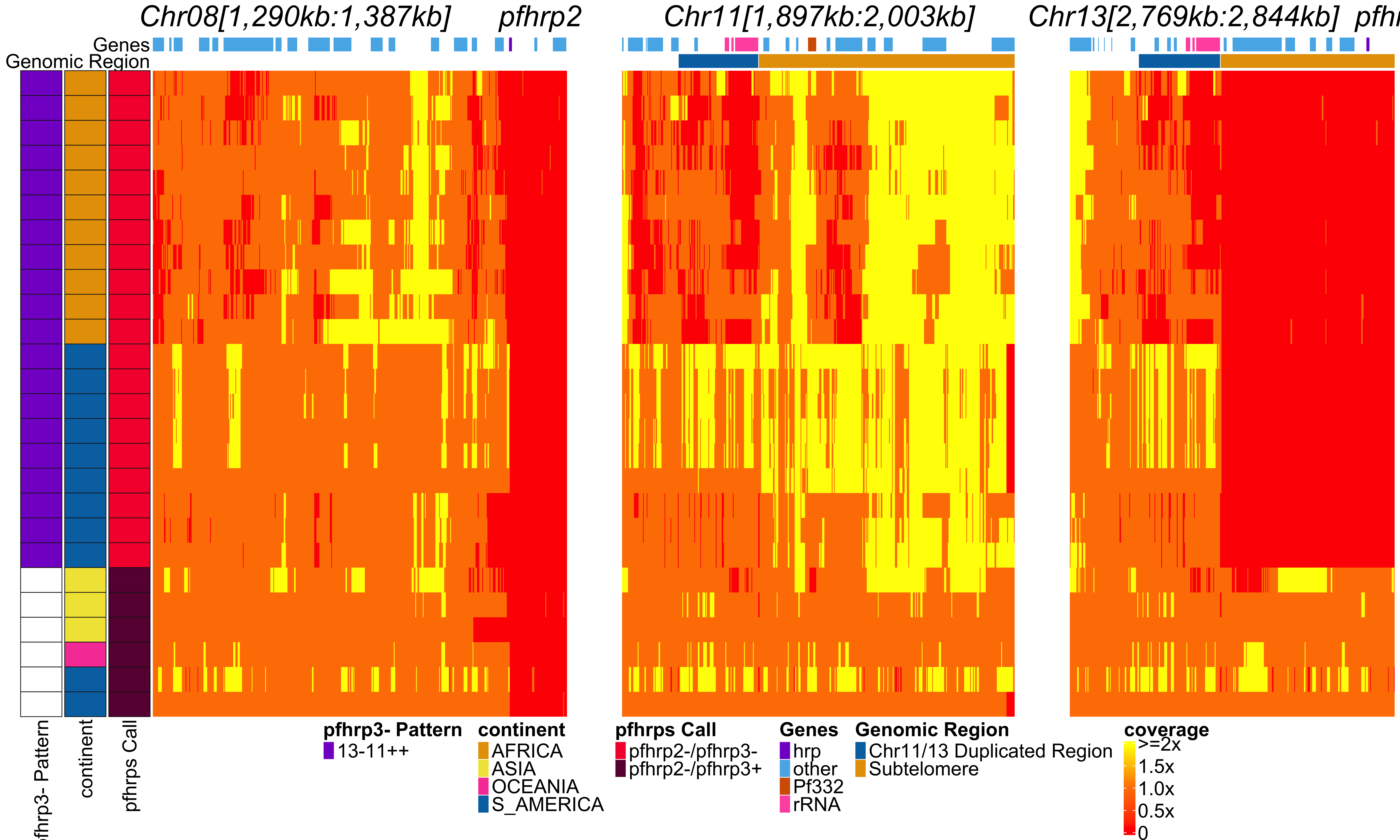
dev.off()Coverage of genomic regions in pfhrp2 deleted strains
Displaying from Pf3D7 genomic version=2015-06-18 of the following chromosome regions
- chromosome 08: 1290365 - 1387982 1,290kb:1,387kb
- chromosome 11: 1897157 - 2003328 1,897kb:2,003kb
- chromosome 13: 2769916 - 2844777 2,769kb:2,844kb
Code
allBasicInfo_filt_sp_mat_hrp2_deleted = allBasicInfo_filt_sp_mat[rownames(allBasicInfo_filt_sp_mat) %in% allMetaDeletionCalls_hrp2_deleted$sample, ]
metaSelected_hrp2_deleted = allMetaDeletionCalls_hrp2_deleted[match(rownames(allBasicInfo_filt_sp_mat_hrp2_deleted), allMetaDeletionCalls_hrp2_deleted$sample), ]
allBasicInfo_filt_sp_mat_hrp2_deleted_hclust = hclust(dist(allBasicInfo_filt_sp_mat_hrp2_deleted))
allBasicInfo_filt_sp_mat_hrp2_deleted_hclust_ordering = tibble(
sample = rownames(allBasicInfo_filt_sp_mat_hrp2_deleted)[allBasicInfo_filt_sp_mat_hrp2_deleted_hclust$order],
hclustInputOrder = allBasicInfo_filt_sp_mat_hrp2_deleted_hclust$order,
hclustOutputOrder = 1:nrow(allBasicInfo_filt_sp_mat_hrp2_deleted)
)
rowAnnoDf_hrp2_deleted = metaSelected_hrp2_deleted %>%
left_join(allBasicInfo_filt_sp_mat_hrp2_deleted_hclust_ordering) %>%
select("Pattern", "secondaryRegion", "hrpCall", hclustOutputOrder) %>%
rename(continent = secondaryRegion) %>%
as.data.frame()
rowAnnoColorsMod = rowAnnoColors
rowAnnoColorsMod[["Pattern"]] = rowAnnoColorsMod_hrp3DeletionPattern
rowAnnoColorsMod[["pfhrp3- Pattern"]] = rowAnnoColorsMod_hrp3DeletionPattern
rowAnnoColorsMod[["DeletionPattern"]] = rowAnnoColorsMod_hrp3DeletionPattern
rowAnnoColorsMod[["pfhrps Call"]] = pfhrpsCallColors
topAnnoColors_mod = list()
topAnnoColors_mod[["Genes"]] = c("other" = "#56B4E9", hrp = "#009E73", Pf332 = "#D55E00", rRNA = "#FF5AAF", "pfmdr1" = "#7CFFFA")
topAnnoColors_mod[["Genes"]] = c("other" = "#56B4E9", hrp = "#8400CD", Pf332 = "#D55E00", rRNA = "#FF5AAF", "pfmdr1" = "#7CFFFA")
topAnnoColors_mod[["Genomic Region"]] = c("Chr11/13 Duplicated Region" = "#0072B2", "Subtelomere" = "#E69F00", "Chr5 Duplication" = "#EF0096")
rowAnnoDf_hrp2_deleted_sorted = rowAnnoDf_hrp2_deleted %>%
as_tibble() %>%
mutate(rowID = row_number()) %>%
# mutate(Pattern = factor(Pattern, levels = c("11++/13-", "5++/13-", "11/13-TARE1", "11-TARE1/13"))) %>%
# mutate(Pattern = factor(Pattern, levels = c("13-11++", "13-5++", "13-TARE1", "13-"))) %>%
mutate(Pattern = factor(Pattern, levels = c("13-11++"))) %>%
arrange(Pattern, continent, hrpCall, hclustOutputOrder) %>%
rename("pfhrps Call" = hrpCall)
rowAnnoDf_hrp2_deleted_sortedDf = rowAnnoDf_hrp2_deleted_sorted %>%
select(-rowID,-hclustOutputOrder) %>%
as.data.frame()
allBasicInfo_filt_sp_mat_noLabs = allBasicInfo_filt_sp_mat_hrp2_deleted
rownames(allBasicInfo_filt_sp_mat_noLabs) = NULL
colnames(allBasicInfo_filt_sp_mat_noLabs) = NULL
allBasicInfo_filt_sp_mat_noLabs_chr08 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_08_", colnames(allBasicInfo_filt_sp_mat))] #08: 1290365 - 1387982 1,290kb:1,387kb
allBasicInfo_filt_sp_mat_noLabs_chr11 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_11_", colnames(allBasicInfo_filt_sp_mat))] #11: 1897157 - 2003328 1,897kb:2,003kb
allBasicInfo_filt_sp_mat_noLabs_chr13 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_13_", colnames(allBasicInfo_filt_sp_mat))] #13: 2769916 - 2844777 2,769kb:2,844kb
#08: 1290365 - 1387982 1,290kb:1,387kb
#11: 1897157 - 2003328 1,897kb:2,003kb
#13: 2769916 - 2844777 2,769kb:2,844kb
allBasicInfo_filt_sp_mat_noLabs_chr08_sorted = allBasicInfo_filt_sp_mat_noLabs_chr08[rowAnnoDf_hrp2_deleted_sorted$rowID,]
allBasicInfo_filt_sp_mat_noLabs_chr11_sorted = allBasicInfo_filt_sp_mat_noLabs_chr11[rowAnnoDf_hrp2_deleted_sorted$rowID,]
allBasicInfo_filt_sp_mat_noLabs_chr13_sorted = allBasicInfo_filt_sp_mat_noLabs_chr13[rowAnnoDf_hrp2_deleted_sorted$rowID,]
rowAnnoDf_hrp2_deleted_sortedDf_mod = rowAnnoDf_hrp2_deleted_sortedDf
rowAnnoDf_hrp2_deleted_sortedDf_mod = rowAnnoDf_hrp2_deleted_sortedDf_mod%>%
rename("pfhrp3- Pattern" = "Pattern")
annotationTextSize = 20
sideAnno = rowAnnotation(
df = rowAnnoDf_hrp2_deleted_sortedDf_mod,
col = rowAnnoColorsMod,
gp = gpar(col = "grey10"),
simple_anno_size = unit(1.5, "cm"),
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
na_col = c("#99999900"),
gap = unit(0.1, "cm")
)
topAnnoDf = regions[,c("inGene", "genomicRegion", "chrom")] %>% as.data.frame()
topAnnoDf_chr08 = topAnnoDf %>% filter(grepl("_08_",chrom)) %>% select(-chrom)
topAnnoDf_chr11 = topAnnoDf %>% filter(grepl("_11_",chrom)) %>% select(-chrom)
topAnnoDf_chr13 = topAnnoDf %>% filter(grepl("_13_",chrom)) %>% select(-chrom)
topAnnoDf_chr08_mod = topAnnoDf_chr08 %>%
rename("Genomic Region" = genomicRegion, "Genes" = "inGene") %>%
mutate(Genes = ifelse(Genes, "other", NA)) %>%
mutate(Genes = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "rRNA"), `Genomic Region`, Genes)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "After Duplicated Region"), "Subtelomere", `Genomic Region`))%>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("rRNA", "Duplicated Region"), "Chr11/13 Duplicated Region", `Genomic Region`))%>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("other"), NA, `Genomic Region`)) %>%
mutate(`Genomic Region` = NA)
topAnnoDf_chr11_mod = topAnnoDf_chr11 %>%
rename("Genomic Region" = genomicRegion, "Genes" = "inGene") %>%
mutate(Genes = ifelse(Genes, "other", NA)) %>%
mutate(Genes = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "rRNA"), `Genomic Region`, Genes)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "After Duplicated Region"), "Subtelomere", `Genomic Region`))%>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("rRNA", "Duplicated Region"), "Chr11/13 Duplicated Region", `Genomic Region`)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("other"), NA, `Genomic Region`))
topAnnoDf_chr13_mod = topAnnoDf_chr13 %>%
rename("Genomic Region" = genomicRegion, "Genes" = "inGene")%>%
mutate(Genes = ifelse(Genes, "other", NA)) %>%
mutate(Genes = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "rRNA"), `Genomic Region`, Genes)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "After Duplicated Region"), "Subtelomere", `Genomic Region`))%>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("rRNA", "Duplicated Region"), "Chr11/13 Duplicated Region", `Genomic Region`)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("other"), NA, `Genomic Region`))
topAnno_chr08 = HeatmapAnnotation(
df = topAnnoDf_chr08_mod,
col = topAnnoColors_mod,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
show_annotation_name = T,
na_col = "#FFFFFF00",
gap = unit(0.1, "cm")
)
topAnno_chr11 = HeatmapAnnotation(
df = topAnnoDf_chr11_mod,
col = topAnnoColors_mod,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
show_annotation_name = F,
na_col = "#FFFFFF00",
gap = unit(0.1, "cm")
)
topAnno_chr13 = HeatmapAnnotation(
df = topAnnoDf_chr13_mod,
col = topAnnoColors_mod,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
show_annotation_name = F,
na_col = "#FFFFFF00",
gap = unit(0.1, "cm")
)
library(circlize)
col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3)))
allCoverageHeatMap_chr08 = Heatmap(
allBasicInfo_filt_sp_mat_noLabs_chr08_sorted,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno_chr08,
left_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
title = "coverage", at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
column_title = "Chr08[1,290kb:1,387kb] pfhrp2",
column_title_gp = gpar(fontsize = 30, fontface = "italic")
)
allCoverageHeatMap_chr11 = Heatmap(
allBasicInfo_filt_sp_mat_noLabs_chr11_sorted,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno_chr11,
#left_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
title = "coverage", at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
column_title = "Chr11[1,897kb:2,003kb]",
column_title_gp = gpar(fontsize = 30, fontface = "italic")
)
allCoverageHeatMap_chr13 = Heatmap(
allBasicInfo_filt_sp_mat_noLabs_chr13_sorted,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno_chr13,
# left_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold",
title = "coverage", at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x"))
),
column_title = "Chr13[2,769kb:2,844kb] pfhrp3",
column_title_gp = gpar(fontsize = 30, fontface = "italic")
)Code
#08: 1290365 - 1387982 1,290kb:1,387kb
#11: 1897157 - 2003328 1,897kb:2,003kb
#13: 2769916 - 2844777 2,769kb:2,844kb
hms_list = allCoverageHeatMap_chr08 + allCoverageHeatMap_chr11 + allCoverageHeatMap_chr13
draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom",
ht_gap = unit(2, "cm"),
#padding = unit(c(5.5 *5*2, 5.5, 5.5, 5.5), "points")
)
Code
#08: 1290365 - 1387982 1,290kb:1,387kb
#11: 1897157 - 2003328 1,897kb:2,003kb
#13: 2769916 - 2844777 2,769kb:2,844kb
pdf("allHeatmap_byChrom_filtered_chr08_chr11_chr13_sorted_hrp2_deleted.pdf", useDingbats = F, width = 20, height = 12)
draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom",
ht_gap = unit(2, "cm"),
padding = unit(c(5.5 *5, 5.5, 5.5, 5.5 *5*2.5), "points")
)
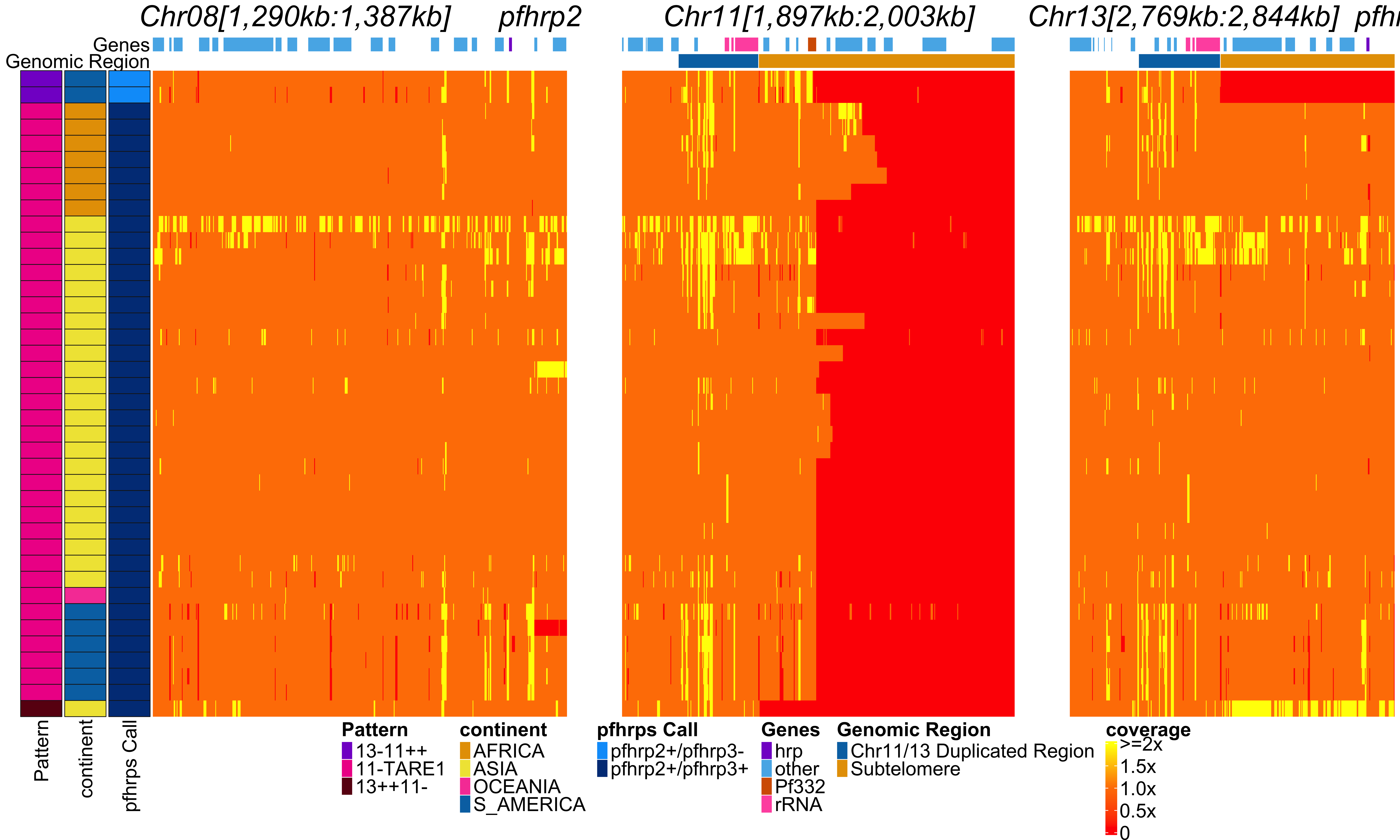
dev.off()Coverage of genomic regions in chromosome 11 deleted strains
Displaying from Pf3D7 genomic version=2015-06-18 of the following chromosome regions
- chromosome 08: 1290365 - 1387982 1,290kb:1,387kb
- chromosome 11: 1897157 - 2003328 1,897kb:2,003kb
- chromosome 13: 2769916 - 2844777 2,769kb:2,844kb
Code
allBasicInfo_filt_sp_mat_chr11_deleted = allBasicInfo_filt_sp_mat[rownames(allBasicInfo_filt_sp_mat) %in% allMetaDeletionCalls_chr11_deleted$sample, ]
metaSelected_chr11_deleted = allMetaDeletionCalls_chr11_deleted[match(rownames(allBasicInfo_filt_sp_mat_chr11_deleted), allMetaDeletionCalls_chr11_deleted$sample), ]
allBasicInfo_filt_sp_mat_chr11_deleted_hclust = hclust(dist(allBasicInfo_filt_sp_mat_chr11_deleted))
allBasicInfo_filt_sp_mat_chr11_deleted_hclust_ordering = tibble(
sample = rownames(allBasicInfo_filt_sp_mat_chr11_deleted)[allBasicInfo_filt_sp_mat_chr11_deleted_hclust$order],
hclustInputOrder = allBasicInfo_filt_sp_mat_chr11_deleted_hclust$order,
hclustOutputOrder = 1:nrow(allBasicInfo_filt_sp_mat_chr11_deleted)
)
rowAnnoDf_chr11_deleted = metaSelected_chr11_deleted %>%
left_join(allBasicInfo_filt_sp_mat_chr11_deleted_hclust_ordering) %>%
select("Pattern", "secondaryRegion", "hrpCall", hclustOutputOrder) %>%
rename(continent = secondaryRegion) %>%
as.data.frame()
rowAnnoColorsMod = rowAnnoColors
rowAnnoColorsMod[["Pattern"]] = rowAnnoColorsMod_hrp3DeletionPattern
rowAnnoColorsMod[["pfhrp3- Pattern"]] = rowAnnoColorsMod_hrp3DeletionPattern
rowAnnoColorsMod[["DeletionPattern"]] = rowAnnoColorsMod_hrp3DeletionPattern
rowAnnoColorsMod[["pfhrps Call"]] = pfhrpsCallColors
topAnnoColors_mod = list()
topAnnoColors_mod[["Genes"]] = c("other" = "#56B4E9", hrp = "#009E73", Pf332 = "#D55E00", rRNA = "#FF5AAF", "pfmdr1" = "#7CFFFA")
topAnnoColors_mod[["Genes"]] = c("other" = "#56B4E9", hrp = "#8400CD", Pf332 = "#D55E00", rRNA = "#FF5AAF", "pfmdr1" = "#7CFFFA")
topAnnoColors_mod[["Genomic Region"]] = c("Chr11/13 Duplicated Region" = "#0072B2", "Subtelomere" = "#E69F00", "Chr5 Duplication" = "#EF0096")
rowAnnoDf_chr11_deleted_sorted = rowAnnoDf_chr11_deleted %>%
as_tibble() %>%
mutate(rowID = row_number()) %>%
# mutate(Pattern = factor(Pattern, levels = c("11++/13-", "5++/13-", "11/13-TARE1", "11-TARE1/13"))) %>%
mutate(Pattern = ifelse(is.na(Pattern), "13++11-", Pattern)) %>%
mutate(Pattern = factor(Pattern, levels = c("13-11++", "11-TARE1", "13++11-"))) %>%
arrange(Pattern, continent, hrpCall, hclustOutputOrder) %>%
#arrange(Pattern, continent, hrpCall) %>%
rename("pfhrps Call" = hrpCall) %>%
select(-hclustOutputOrder)
rowAnnoDf_chr11_deleted_sortedDf = rowAnnoDf_chr11_deleted_sorted %>%
select(-rowID) %>%
as.data.frame()
allBasicInfo_filt_sp_mat_noLabs = allBasicInfo_filt_sp_mat_chr11_deleted
rownames(allBasicInfo_filt_sp_mat_noLabs) = NULL
colnames(allBasicInfo_filt_sp_mat_noLabs) = NULL
allBasicInfo_filt_sp_mat_noLabs_chr08 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_08_", colnames(allBasicInfo_filt_sp_mat))] #08: 1290365 - 1387982 1,290kb:1,387kb
allBasicInfo_filt_sp_mat_noLabs_chr11 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_11_", colnames(allBasicInfo_filt_sp_mat))] #11: 1897157 - 2003328 1,897kb:2,003kb
allBasicInfo_filt_sp_mat_noLabs_chr13 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_13_", colnames(allBasicInfo_filt_sp_mat))] #13: 2769916 - 2844777 2,769kb:2,844kb
#08: 1290365 - 1387982 1,290kb:1,387kb
#11: 1897157 - 2003328 1,897kb:2,003kb
#13: 2769916 - 2844777 2,769kb:2,844kb
allBasicInfo_filt_sp_mat_noLabs_chr08_sorted = allBasicInfo_filt_sp_mat_noLabs_chr08[rowAnnoDf_chr11_deleted_sorted$rowID,]
allBasicInfo_filt_sp_mat_noLabs_chr11_sorted = allBasicInfo_filt_sp_mat_noLabs_chr11[rowAnnoDf_chr11_deleted_sorted$rowID,]
allBasicInfo_filt_sp_mat_noLabs_chr13_sorted = allBasicInfo_filt_sp_mat_noLabs_chr13[rowAnnoDf_chr11_deleted_sorted$rowID,]
rowAnnoDf_chr11_deleted_sortedDf_mod = rowAnnoDf_chr11_deleted_sortedDf
rowAnnoDf_chr11_deleted_sortedDf_mod = rowAnnoDf_chr11_deleted_sortedDf_mod%>%
rename("pfhrp3- Pattern" = "Pattern")
annotationTextSize = 20
sideAnno = rowAnnotation(
df = rowAnnoDf_chr11_deleted_sortedDf,
col = rowAnnoColorsMod,
gp = gpar(col = "grey10"),
simple_anno_size = unit(1.5, "cm"),
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
na_col = c("#99999900"),
gap = unit(0.1, "cm")
)
topAnnoDf = regions[,c("inGene", "genomicRegion", "chrom")] %>% as.data.frame()
topAnnoDf_chr08 = topAnnoDf %>% filter(grepl("_08_",chrom)) %>% select(-chrom)
topAnnoDf_chr11 = topAnnoDf %>% filter(grepl("_11_",chrom)) %>% select(-chrom)
topAnnoDf_chr13 = topAnnoDf %>% filter(grepl("_13_",chrom)) %>% select(-chrom)
topAnnoDf_chr08_mod = topAnnoDf_chr08 %>%
rename("Genomic Region" = genomicRegion, "Genes" = "inGene") %>%
mutate(Genes = ifelse(Genes, "other", NA)) %>%
mutate(Genes = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "rRNA"), `Genomic Region`, Genes)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "After Duplicated Region"), "Subtelomere", `Genomic Region`))%>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("rRNA", "Duplicated Region"), "Chr11/13 Duplicated Region", `Genomic Region`))%>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("other"), NA, `Genomic Region`)) %>%
mutate(`Genomic Region` = NA)
topAnnoDf_chr11_mod = topAnnoDf_chr11 %>%
rename("Genomic Region" = genomicRegion, "Genes" = "inGene") %>%
mutate(Genes = ifelse(Genes, "other", NA)) %>%
mutate(Genes = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "rRNA"), `Genomic Region`, Genes)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "After Duplicated Region"), "Subtelomere", `Genomic Region`))%>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("rRNA", "Duplicated Region"), "Chr11/13 Duplicated Region", `Genomic Region`)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("other"), NA, `Genomic Region`))
topAnnoDf_chr13_mod = topAnnoDf_chr13 %>%
rename("Genomic Region" = genomicRegion, "Genes" = "inGene")%>%
mutate(Genes = ifelse(Genes, "other", NA)) %>%
mutate(Genes = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "rRNA"), `Genomic Region`, Genes)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "After Duplicated Region"), "Subtelomere", `Genomic Region`))%>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("rRNA", "Duplicated Region"), "Chr11/13 Duplicated Region", `Genomic Region`)) %>%
mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("other"), NA, `Genomic Region`))
topAnno_chr08 = HeatmapAnnotation(
df = topAnnoDf_chr08_mod,
col = topAnnoColors_mod,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
show_annotation_name = T,
na_col = "#FFFFFF00",
gap = unit(0.1, "cm")
)
topAnno_chr11 = HeatmapAnnotation(
df = topAnnoDf_chr11_mod,
col = topAnnoColors_mod,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
show_annotation_name = F,
na_col = "#FFFFFF00",
gap = unit(0.1, "cm")
)
topAnno_chr13 = HeatmapAnnotation(
df = topAnnoDf_chr13_mod,
col = topAnnoColors_mod,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
show_annotation_name = F,
na_col = "#FFFFFF00",
gap = unit(0.1, "cm")
)
library(circlize)
col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3)))
allCoverageHeatMap_chr08 = Heatmap(
allBasicInfo_filt_sp_mat_noLabs_chr08_sorted,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno_chr08,
left_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
title = "coverage", at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
column_title = "Chr08[1,290kb:1,387kb] pfhrp2",
column_title_gp = gpar(fontsize = 30, fontface = "italic")
)
allCoverageHeatMap_chr11 = Heatmap(
allBasicInfo_filt_sp_mat_noLabs_chr11_sorted,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno_chr11,
#left_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
title = "coverage", at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
column_title = "Chr11[1,897kb:2,003kb]",
column_title_gp = gpar(fontsize = 30, fontface = "italic")
)
allCoverageHeatMap_chr13 = Heatmap(
allBasicInfo_filt_sp_mat_noLabs_chr13_sorted,
cluster_columns = F,
cluster_rows = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno_chr13,
# left_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold",
title = "coverage", at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x"))
),
column_title = "Chr13[2,769kb:2,844kb] pfhrp3",
column_title_gp = gpar(fontsize = 30, fontface = "italic")
)Code
#08: 1290365 - 1387982 1,290kb:1,387kb
#11: 1897157 - 2003328 1,897kb:2,003kb
#13: 2769916 - 2844777 2,769kb:2,844kb
hms_list = allCoverageHeatMap_chr08 + allCoverageHeatMap_chr11 + allCoverageHeatMap_chr13
draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom",
ht_gap = unit(2, "cm"),
#padding = unit(c(5.5 *5*2, 5.5, 5.5, 5.5), "points")
)
Code
#08: 1290365 - 1387982 1,290kb:1,387kb
#11: 1897157 - 2003328 1,897kb:2,003kb
#13: 2769916 - 2844777 2,769kb:2,844kb
pdf("allHeatmap_byChrom_filtered_chr08_chr11_chr13_sorted_chr11_deleted.pdf", useDingbats = F, width = 20, height = 12)
draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom",
ht_gap = unit(2, "cm"),
padding = unit(c(5.5, 5.5, 5.5, 5.5 *5*2.5), "points")
)
dev.off()