Code
quast -r /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta -o quast_PfSD01_flye pilon.fastaEvaluation with the quast(Gurevich et al. 2013) program for comparing genome assemblies
quast -r /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta -o quast_PfSD01_flye pilon.fastaquastTopRes = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/quast_PfSD01_flye/transposed_report.tsv")
create_dt(quastTopRes)Annotating the final contigs using using Companion(Steinbiss et al. 2016)
cd ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/pilon_annotations/PfSD01_11_contig74
elucidator gffToBedByFeature --features polypeptide,rRNA --gff pseudo.out.gff3 --overWrite --extraAttributes product --out geneProducts.bed
sed 's/evidence.*;//g' geneProducts.bed > renamed_geneProducts.bed
elucidator splitColumnContainingMeta --removeEmptyColumn --delim tab --column col.6 --file renamed_geneProducts.bed --addHeader --out renamed_geneProducts.tab.txt --overWritecd ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/pilon_annotations/PfSD01_11_contig74
elucidator gffToBedByFeature --features polypeptide,rRNA --gff pseudo.out.gff3 --overWrite --extraAttributes product --out geneProducts.bed
sed 's/evidence.*;//g' geneProducts.bed > renamed_geneProducts.bed
elucidator splitColumnContainingMeta --removeEmptyColumn --delim tab --column col.6 --file renamed_geneProducts.bed --addHeader --out renamed_geneProducts.tab.txt --overWritecd ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/pilon_annotations
elucidator extractRefSeqsFromGenomes --bed "/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/rRNA/all/merged_updatedrRNA_locs.bed" --genomeDir ../../../../HB3_new_assembly/fakeGenomes// --primaryGenome Pf3D7 --outputDir extract_rRNA_regions --numThreads 15 --overWriteDirs --selectedGenomes Pf3D7,PfSD01_contig_74_pilon,PfSD01_contig_73_chr13_pilon
elucidator extractRefSeqsFromGenomes --bed "/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/rRNA/all/28s_rRNA_locs.bed" --genomeDir ../../../../HB3_new_assembly/fakeGenomes// --primaryGenome Pf3D7 --outputDir extract_28s_rRNA_regions --numThreads 15 --overWriteDirs --selectedGenomes Pf3D7,PfSD01_contig_74_pilon,PfSD01_contig_73_chr13_piloncd ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13
elucidator runTRF --fasta pilon.fasta --supplement --dout trf_pilon --overWriteDir
elucidator extractFromGenomesAndCompare --genomeDir pilon_annotations/ --numThreads 2 --target sharedRegion --sample Pf3D7 --program exact --verbose --dout sharedRegionBlast --fasta ../../../../sharedBetween11_and_13/investigatingChrom11Chrom13/shared_11_13_region.fasta --overWriteDir
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 pilon_annotations/contig_74_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_11_v3.fasta --out PfSD01_11_vs_Pf3D7_11_klen19 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 pilon_annotations/contig_74_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_13_v3.fasta --out PfSD01_11_vs_Pf3D7_13_klen19 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 pilon_annotations/contig_73_chr13_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_11_v3.fasta --out PfSD01_13_vs_Pf3D7_11_klen19 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 pilon_annotations/contig_73_chr13_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_13_v3.fasta --out PfSD01_13_vs_Pf3D7_13_klen19 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 pilon_annotations/contig_74_pilon.fasta --seq2 pilon_annotations/contig_73_chr13_pilon.fasta --out SD01_11_vs_SD01_13_klen19 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 pilon_annotations/contig_73_chr13_pilon.fasta --seq2 pilon_annotations/contig_74_pilon.fasta --out SD01_13_vs_SD01_11_klen19 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 35 --seq1 pilon_annotations/contig_74_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_11_v3.fasta --out PfSD01_11_vs_Pf3D7_11_klen35 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 35 --seq1 pilon_annotations/contig_74_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_13_v3.fasta --out PfSD01_11_vs_Pf3D7_13_klen35 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 35 --seq1 pilon_annotations/contig_73_chr13_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_11_v3.fasta --out PfSD01_13_vs_Pf3D7_11_klen35 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 35 --seq1 pilon_annotations/contig_73_chr13_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_13_v3.fasta --out PfSD01_13_vs_Pf3D7_13_klen35 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 35 --seq1 pilon_annotations/contig_74_pilon.fasta --seq2 pilon_annotations/contig_73_chr13_pilon.fasta --out SD01_11_vs_SD01_13_klen35 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 35 --seq1 pilon_annotations/contig_73_chr13_pilon.fasta --seq2 pilon_annotations/contig_74_pilon.fasta --out SD01_13_vs_SD01_11_klen35 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 pilon_annotations/contig_74_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_11_v3.fasta --out PfSD01_11_vs_Pf3D7_11_klen300 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 pilon_annotations/contig_74_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_13_v3.fasta --out PfSD01_11_vs_Pf3D7_13_klen300 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 pilon_annotations/contig_73_chr13_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_11_v3.fasta --out PfSD01_13_vs_Pf3D7_11_klen300 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 pilon_annotations/contig_73_chr13_pilon.fasta --seq2 ../../../HB3_new_assembly/fakeGenomes/Pf3D7_13_v3.fasta --out PfSD01_13_vs_Pf3D7_13_klen300 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 pilon_annotations/contig_74_pilon.fasta --seq2 pilon_annotations/contig_73_chr13_pilon.fasta --out SD01_11_vs_SD01_13_klen300 --collapse --overWrite
elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 pilon_annotations/contig_73_chr13_pilon.fasta --seq2 pilon_annotations/contig_74_pilon.fasta --out SD01_13_vs_SD01_11_klen300 --collapse --overWrite
# finding all exact matches regardless of uniqueness
mummer -b -c -F -l 200 -maxmatch pilon_annotations/contig_74_pilon.fasta ../../../HB3_new_assembly/fakeGenomes/Pf3D7_11_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfSD01_11_vs_Pf3D7_11_exactMatches_minlen200.tab.txt
mummer -b -c -F -l 200 -maxmatch pilon_annotations/contig_74_pilon.fasta ../../../HB3_new_assembly/fakeGenomes/Pf3D7_13_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfSD01_11_vs_Pf3D7_13_exactMatches_minlen200.tab.txt
mummer -b -c -F -l 200 -maxmatch pilon_annotations/contig_73_chr13_pilon.fasta ../../../HB3_new_assembly/fakeGenomes/Pf3D7_11_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfSD01_13_vs_Pf3D7_11_exactMatches_minlen200.tab.txt
mummer -b -c -F -l 200 -maxmatch pilon_annotations/contig_73_chr13_pilon.fasta ../../../HB3_new_assembly/fakeGenomes/Pf3D7_13_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfSD01_13_vs_Pf3D7_13_exactMatches_minlen200.tab.txt
mummer -b -c -F -l 200 -maxmatch pilon_annotations/contig_74_pilon.fasta pilon_annotations/contig_73_chr13_pilon.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > SD01_11_vs_SD01_13_exactMatches_minlen200.tab.txt
mummer -b -c -F -l 200 -maxmatch pilon_annotations/contig_73_chr13_pilon.fasta pilon_annotations/contig_74_pilon.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > SD01_13_vs_SD01_11_exactMatches_minlen200.tab.txt
elucidator getReadLens --fasta pilon.fasta --trimAtWhiteSpace > chroms_pilon.txtlastz contig_74_pilon.fasta contig_73_chr13_pilon.fasta --format=general --output=contig74_vs_contig73.tsvrRNA_28s = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/pilon_annotations/extract_28s_rRNA_regions/Pf3D7_11_v3-1928933-1933138-for/beds/PfSD01_contig_73_chr13_pilon_bestRegion.bed", col_names = F) %>%
bind_rows(readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/pilon_annotations/extract_28s_rRNA_regions/Pf3D7_11_v3-1928933-1933138-for/beds/PfSD01_contig_74_pilon_bestRegion.bed", col_names = F))%>%
rename(col.0 = X1,
col.1 = X2,
col.2 = X3,
col.3 = X4,
col.4 = X5,
col.5 = X6) %>%
mutate(ID = "PfSD01_rRNA-28s", feature = "rRNA", product = "rRNA", product_mod = "rRNA")
annotations = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/pilon_annotations/PfSD01_11_contig74/renamed_geneProducts.tab.txt") %>%
bind_rows(readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/pilon_annotations/PfSD01_13_contig73/renamed_geneProducts.tab.txt") ) %>%
mutate(col.0 = gsub("PfSD01_11_contig74_11", "contig_74_pilon", col.0))%>%
mutate(col.0 = gsub("PfSD01_13_contig73_13", "contig_73_chr13_pilon", col.0))
annotations = annotations %>%
mutate(product_mod = gsub("term=", "", product)) %>%
#rename(chrom = col.0, start = col.1, end = col.2, name = col.3, length = col.4, strand = col.5) %>%
# mutate(gene = ifelse(grepl("PF3D7_0831800", product_mod), "HRP II", "other")) %>%
# mutate(gene = ifelse(grepl("PF3D7_1372200", product_mod), "HRP III", gene)) %>%
mutate(product_mod = gsub("unknownfunction", "unknown function", product_mod)) %>%
mutate(product_mod = gsub(" \\(SURFIN", "\\(SURFIN", product_mod)) %>%
mutate(product_mod = ifelse(grepl("Plasmodium exported protein", product_mod), "Plasmodium exported protein (PHIST)", product_mod)) %>%
mutate(product_mod = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==product_mod, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", product_mod)) %>%
mutate(product_mod = ifelse("membrane associated erythrocyte binding-like protein"==product_mod, "merozoite adhesive erythrocytic binding protein", product_mod)) %>%
mutate(product_mod = ifelse("membrane associated erythrocyte binding-likeprotein"==product_mod, "merozoite adhesive erythrocytic binding protein", product_mod)) %>%
mutate(product_mod = ifelse("membrane associated histidine-rich protein 1"==product_mod, "membrane associated histidine-rich protein", product_mod)) %>%
mutate(product_mod = gsub(",putative", ", putative", product_mod)) %>%
mutate(product_mod = gsub(",pseudogene", ", pseudogene", product_mod))%>%
mutate(product_mod = gsub("unknownfunction", "unknown function", product_mod))%>%
mutate(product_mod = gsub("conserved protein, unknown function", "conserved Plasmodium protein, unknown function", product_mod)) %>%
mutate(product_mod = gsub(" \\(SURFIN", "\\(SURFIN", product_mod)) %>%
mutate(product_mod = ifelse(grepl("Plasmodium exported protein", product_mod), "Plasmodium exported protein (PHIST)", product_mod)) %>%
mutate(product_mod = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==product_mod, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", product_mod)) %>%
mutate(product_mod = ifelse("membrane associated histidine-rich protein 1"==product_mod, "membrane associated histidine-rich protein", product_mod))%>%
mutate(product_mod = gsub(",pseudogene", ", pseudogene", product_mod))%>%
mutate(product_mod = gsub("unknownfunction", "unknown function", product_mod))%>%
mutate(product_mod = gsub(" \\(SURFIN", "\\(SURFIN", product_mod)) %>%
mutate(product_mod = ifelse(grepl("Plasmodium exported protein", product_mod), "Plasmodium exported protein (PHIST)", product_mod)) %>%
mutate(product_mod = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==product_mod, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", product_mod)) %>%
mutate(product_mod = ifelse(grepl("sporozoite and liver stage tryptophan-rich protein, putative", product_mod), "tryptophan/threonine-rich antigen", product_mod))%>%
mutate(product_mod = ifelse(grepl("CRA domain-containing protein, putative", product_mod), "conserved Plasmodium protein, unknown function", product_mod))%>%
mutate(product_mod = gsub(",pseudogene", ", pseudogene", product_mod)) %>%
mutate(product_mod = gsub("surfaceantigen", "surface antigen", product_mod)) %>%
mutate(product_mod = gsub("Tetratricopeptide repeat, putative", "tetratricopeptide repeat protein, putative", product_mod)) %>%
mutate(product_mod = gsub("transmembraneprotein", "transmembrane protein", product_mod)) %>%
mutate(product_mod = ifelse(grepl("PfEMP1", product_mod) & grepl("pseudogene", product_mod), "erythrocyte membrane protein 1 (PfEMP1), pseudogene", product_mod)) %>%
mutate(product_mod = gsub("PIR protein", "stevor", product_mod)) %>%
mutate(product_mod = gsub("erythrocyte membrane protein 1-like", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", product_mod)) %>%
mutate(product_mod = gsub("acidic terminal segments, variant surface antigen of PfEMP1, putative", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", product_mod))%>%
mutate(product_mod = ifelse(grepl("CoA binding protein", product_mod, ignore.case = T), "acyl-CoA binding protein", product_mod)) %>%
mutate(product_mod = ifelse(grepl("transfer RNA", product_mod) | grepl("tRNA", product_mod), "tRNA", product_mod))%>%
mutate(product_mod = ifelse(grepl("cytoadherence", product_mod), "CLAG", product_mod))%>%
mutate(product_mod = ifelse(grepl("surface-associated interspersed protein", product_mod), "SURFIN", product_mod))%>%
mutate(product_mod = ifelse(grepl("SURFIN", product_mod), "SURFIN", product_mod))%>%
mutate(product_mod = ifelse(grepl("stevor-like", product_mod), "stevor, pseudogene", product_mod)) %>%
mutate(product_mod = ifelse(grepl("exported protein family", product_mod), "exported protein family", product_mod)) %>%
mutate(product_mod = ifelse(grepl("rRNA", feature), "rRNA", product_mod)) %>%
mutate(product_mod = ifelse(grepl("serine/threonine protein kinase", product_mod), "serine/threonine protein kinase, FIKK family", product_mod)) %>%
mutate(product_mod = ifelse(grepl("hypothetical protein", product_mod), "hypothetical protein, conserved", product_mod)) %>%
mutate(product_mod = ifelse(grepl("conserved Plasmodium protein, unknown function", product_mod), "hypothetical protein, conserved", product_mod)) %>%
mutate(product_mod = ifelse(grepl("Rifin/stevor family, putative", product_mod), "stevor", product_mod)) %>%
mutate(product_mod = ifelse(grepl("erythrocyte membrane protein 1 (PfEMP1), pseudogene", product_mod), "erythrocyte membrane protein 1 (PfEMP1)", product_mod)) %>%
mutate(product_mod = ifelse(grepl("erythrocyte membrane protein 1", product_mod), "erythrocyte membrane protein 1 (PfEMP1)", product_mod))%>%
mutate(product_mod = ifelse(grepl("Plasmodium RESA N-terminal, putative", product_mod), "ring-infected erythrocyte surface antigen", product_mod)) %>%
left_join(rRNA_28s %>%
select(col.0, col.1, col.2) %>%
rename(`28rRNA-start` = col.1,
`28rRNA-end` = col.2)) %>%
filter(!(col.1 >= `28rRNA-start` & col.1 <= `28rRNA-end`)) %>%
bind_rows(rRNA_28s)sharedRegion = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/sharedRegionBlast/contig_73_chr13_pilon/regions.bed", col_names = F) %>%
bind_rows(
readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/sharedRegionBlast/contig_74_pilon/regions.bed", col_names = F)
) %>%
unique() %>%
mutate(col.0 = X1,
col.1 = X2,
col.2 = X3,
start = X2,
end = X3)
chromLengths = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/chroms_pilon.txt", col_names = c("col.0", "chromLength"))
backtrace = 30000-500
rRNA_28s_forLimiting = rRNA_28s %>%
mutate(filtStart = col.1 - backtrace) %>%
select(col.0, filtStart) %>%
left_join(chromLengths)
annotations_filt = annotations %>%
left_join(rRNA_28s_forLimiting) %>%
filter(col.1 >= filtStart)
rawGeneAnnotationsColors = scheme$hex(length(unique(c(annotations_filt$product_mod))))
names(rawGeneAnnotationsColors) = unique(c(annotations_filt$product_mod))
rawGeneAnnotationsColorsFromHB3 = readr::read_tsv("../HB3_new_assembly/rawGeneAnnotationsColors.tab.txt")
rawGeneAnnotationsColors = rawGeneAnnotationsColorsFromHB3$colors
names(rawGeneAnnotationsColors) = rawGeneAnnotationsColorsFromHB3$names
genomicElementsColors = c()
genomicElementsColors["Duplicated\nRegion"] = "#2271B2"
genomicElementsColors["Exact Matches\nSD01-chr11-to-chr13"] = "#F748A5"
genomicElementsColors["Exact Matches\n3D7 chr11"] = "#3DB7E9"
# genomicElementsColors["Telomere Associated\nRepetitive Element"] = "#D55E00"
# genomicElementsColors["Duplicated\nRegion"] = "#4E79A7"
# genomicElementsColors["Exact Matches\nSD01-chr11-to-chr13"] = "#E15759"
# genomicElementsColors["Exact Matches\n3D7 chr11"] = "#59A14F"
# genomicElementsColors["Telomere Associated\nRepetitive Element"] = "#B07AA1"
geneAnnotationsColors = c(rawGeneAnnotationsColors, genomicElementsColors)
annotations_filt = annotations_filt %>%
mutate(col.0 = factor(col.0, levels = c("contig_74_pilon", "contig_73_chr13_pilon")))
rRNA_28s_forLimiting_filer = rRNA_28s_forLimiting %>%
mutate(length = chromLength - filtStart) %>%
mutate(maxLength = max(length)) %>%
mutate(fakeEnd = filtStart + maxLength)
sharedRegion_3d7 = readr::read_tsv("../../sharedBetween11_and_13/investigatingChrom11Chrom13/shared_11_13_region.bed", col_names = T)
# sd01_11_vs_sd01_13 = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/SD01_11_vs_SD01_13_klen19.tab.txt")
# sd01_13_vs_sd01_11 = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/SD01_13_vs_SD01_11_klen19.tab.txt")
# sd01_11_vs_3d7_11 = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/PfSD01_11_vs_Pf3D7_11_klen19.tab.txt")
# sd01_13_vs_3d7_11 = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/PfSD01_13_vs_Pf3D7_11_klen19.tab.txt")
sd01_11_vs_sd01_13_exacts = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/SD01_11_vs_SD01_13_exactMatches_minlen200.tab.txt") %>% filter(strand == "+")
sd01_13_vs_sd01_11_exacts = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/SD01_13_vs_SD01_11_exactMatches_minlen200.tab.txt") %>% filter(strand == "+")
sd01_11_vs_3d7_11_exacts = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/PfSD01_11_vs_Pf3D7_11_exactMatches_minlen200.tab.txt") %>%
left_join(sharedRegion_3d7 %>%
rename(query = `#chrom`, sharedStart = start, sharedEnd = end) %>%
select(query, sharedStart, sharedEnd)) %>%
filter(queryStart >= sharedStart - backtrace)
sd01_13_vs_3d7_11_exacts = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/PfSD01_13_vs_Pf3D7_11_exactMatches_minlen200.tab.txt") %>% filter(strand == "+") %>%
left_join(sharedRegion_3d7 %>%
rename(query = `#chrom`, sharedStart = start, sharedEnd = end) %>%
select(query, sharedStart, sharedEnd)) %>%
filter(queryStart >= sharedStart - backtrace)
sd01_11_vs_sd01_13 = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/SD01_11_vs_SD01_13_klen300.tab.txt")
sd01_13_vs_sd01_11 = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/SD01_13_vs_SD01_11_klen300.tab.txt")
sd01_11_vs_3d7_11 = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/PfSD01_11_vs_Pf3D7_11_klen300.tab.txt")
sd01_13_vs_3d7_11 = readr::read_tsv("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/PfSD01_13_vs_Pf3D7_11_klen300.tab.txt")
sd01_13_vs_sd01_11_forPlotting = bind_rows(
sd01_13_vs_sd01_11 %>%
select(seq1, seq1Pos, size) %>%
rename(col.0 = seq1,
col.1 = seq1Pos) %>%
mutate(col.2 = col.1 + size),
sd01_13_vs_sd01_11 %>%
select(seq2, seq2Pos, size) %>%
rename(col.0 = seq2,
col.1 = seq2Pos) %>%
mutate(col.2 = col.1 + size)
)%>%
left_join(rRNA_28s_forLimiting_filer %>%
select(col.0, filtStart)) %>%
filter(col.1 >= filtStart)%>%
mutate(start = col.1,
end = col.2)
sd01_13_vs_sd01_11_exact_forPlotting = bind_rows(
sd01_13_vs_sd01_11_exacts %>%
select(`#target`, targetStart, length, query, queryStart, queryEnd) %>%
rename(col.0 = `#target`,
col.1 = targetStart,
other = query,
otherStart = queryStart,
otherEnd = queryEnd
) %>%
mutate(col.2 = col.1 + length),
sd01_13_vs_sd01_11_exacts %>%
select(query, queryStart, length, `#target`, targetStart, targetEnd) %>%
rename(col.0 = query,
col.1 = queryStart,
other = `#target`,
otherStart = targetStart,
otherEnd = targetEnd
) %>%
mutate(col.2 = col.1 + length)
)%>%
left_join(rRNA_28s_forLimiting_filer %>%
select(col.0, filtStart)) %>%
filter(col.1 >= filtStart)%>%
mutate(start = col.1,
end = col.2)
sd01_13_and_sd01_11_vs3d7_exacts_forPlotting = bind_rows(
sd01_11_vs_3d7_11_exacts %>%
select(`#target`, targetStart, length, query, queryStart, queryEnd) %>%
rename(col.0 = `#target`,
col.1 = targetStart,
other = query,
otherStart = queryStart,
otherEnd = queryEnd
) %>%
mutate(col.2 = col.1 + length),
sd01_13_vs_3d7_11_exacts %>%
select(`#target`, targetStart, length, query, queryStart, queryEnd) %>%
rename(col.0 = `#target`,
col.1 = targetStart,
other = query,
otherStart = queryStart,
otherEnd = queryEnd
) %>%
mutate(col.2 = col.1 + length)
) %>%
left_join(rRNA_28s_forLimiting_filer %>%
select(col.0, filtStart)) %>%
filter(col.1 >= filtStart)%>%
mutate(start = col.1,
end = col.2)
sd01_13_and_sd01_11_vs3d7_forPlotting = bind_rows(
sd01_11_vs_3d7_11 %>%
select(seq1, seq1Pos, size) %>%
rename(col.0 = seq1,
col.1 = seq1Pos) %>%
mutate(col.2 = col.1 + size),
sd01_13_vs_3d7_11 %>%
select(seq1, seq1Pos, size) %>%
rename(col.0 = seq1,
col.1 = seq1Pos) %>%
mutate(col.2 = col.1 + size)
) %>%
left_join(rRNA_28s_forLimiting_filer %>%
select(col.0, filtStart)) %>%
filter(col.1 >= filtStart)%>%
mutate(start = col.1,
end = col.2)
# rename
rRNA_28s_forLimiting_filer = rRNA_28s_forLimiting_filer %>%
mutate(col.0 = gsub("contig_73_chr13_pilon", "SD01_13", col.0)) %>%
mutate(col.0 = gsub("contig_74_pilon", "SD01_11", col.0))
rRNA_28s_forLimiting = rRNA_28s_forLimiting%>%
mutate(col.0 = gsub("contig_73_chr13_pilon", "SD01_13", col.0)) %>%
mutate(col.0 = gsub("contig_74_pilon", "SD01_11", col.0))
annotations_filt = annotations_filt%>%
mutate(col.0 = gsub("contig_73_chr13_pilon", "SD01_13", col.0)) %>%
mutate(col.0 = gsub("contig_74_pilon", "SD01_11", col.0))
sharedRegion = sharedRegion%>%
mutate(col.0 = gsub("contig_73_chr13_pilon", "SD01_13", col.0)) %>%
mutate(col.0 = gsub("contig_74_pilon", "SD01_11", col.0))
sd01_13_vs_sd01_11_forPlotting = sd01_13_vs_sd01_11_forPlotting%>%
mutate(col.0 = gsub("contig_73_chr13_pilon", "SD01_13", col.0)) %>%
mutate(col.0 = gsub("contig_74_pilon", "SD01_11", col.0))
sd01_13_and_sd01_11_vs3d7_forPlotting = sd01_13_and_sd01_11_vs3d7_forPlotting %>%
mutate(col.0 = gsub("contig_73_chr13_pilon", "SD01_13", col.0)) %>%
mutate(col.0 = gsub("contig_74_pilon", "SD01_11", col.0))
sd01_13_vs_sd01_11_exact_forPlotting = sd01_13_vs_sd01_11_exact_forPlotting %>%
mutate(col.0 = gsub("contig_73_chr13_pilon", "SD01_13", col.0)) %>%
mutate(col.0 = gsub("contig_74_pilon", "SD01_11", col.0))
sd01_13_and_sd01_11_vs3d7_exacts_forPlotting = sd01_13_and_sd01_11_vs3d7_exacts_forPlotting %>%
mutate(col.0 = gsub("contig_73_chr13_pilon", "SD01_13", col.0)) %>%
mutate(col.0 = gsub("contig_74_pilon", "SD01_11", col.0))
sd01_chr11_chr13_annotation_plots = ggplot() +
geom_rect(
aes(
xmin = filtStart,
xmax = fakeEnd,
ymin = 0,
ymax = 1
),
fill = "#FFFFFF00",
data = rRNA_28s_forLimiting_filer
) +
geom_rect(
aes(
xmin = filtStart,
xmax = chromLength,
ymin = 0,
ymax = 1
),
fill = "grey60",
color = "grey60",
data = rRNA_28s_forLimiting
) +
geom_rect(
aes(
xmin = col.1,
xmax = col.2,
ymin = 0,
ymax = 1,
fill = product_mod,
# color = product_mod
), color = "black",
data = annotations_filt
) +
geom_rect(
aes(
xmin = X2,
xmax = X3,
ymin = -0.1,
ymax = 0,
start = start,
end = end,
fill = "Duplicated\nRegion",
color = "Duplicated\nRegion"
),
data = sharedRegion
) +
# geom_rect(
# aes(
# xmin = col.1,
# xmax = col.2,
# ymin = -0.25,
# ymax = -0.1,
# start = start,
# end = end,
# fill = "Exact Matches\nSD01-chr11-to-chr13"
# ),
# data = sd01_13_vs_sd01_11_forPlotting
# ) +
geom_rect(
aes(
xmin = col.1,
xmax = col.2,
ymin = -0.25,
ymax = -0.1,
start = start,
end = end,
length = length,
other = other,
otherStart = otherStart,
otherEnd = otherEnd,
fill = "Exact Matches\nSD01-chr11-to-chr13",
color = "Exact Matches\nSD01-chr11-to-chr13"
),
data = sd01_13_vs_sd01_11_exact_forPlotting
) +
# geom_rect(
# aes(
# xmin = col.1,
# xmax = col.2,
# ymin = -0.40,
# ymax = -0.25,
# start = start,
# end = end,
# fill = "Exact Matches\n3D7 chr11"
# ),
# data = sd01_13_and_sd01_11_vs3d7_forPlotting
# ) +
geom_rect(
aes(
xmin = col.1,
xmax = col.2,
ymin = -0.40,
ymax = -0.25,
start = start,
end = end,
length = length,
other = other,
otherStart = otherStart,
otherEnd = otherEnd,
fill = "Exact Matches\n3D7 chr11",
color = "Exact Matches\n3D7 chr11"
),
data = sd01_13_and_sd01_11_vs3d7_exacts_forPlotting
) +
facet_wrap( ~ col.0,
scales = "free",
ncol = 1,
strip.position = "left") +
sofonias_theme_backgroundTransparent +
theme(axis.text.y = element_blank(),
axis.ticks.y = element_blank()) +
scale_fill_manual(
"Genes",
values = geneAnnotationsColors,
breaks = names(rawGeneAnnotationsColors[unique(annotations_filt$product_mod)]),
labels = names(rawGeneAnnotationsColors[unique(annotations_filt$product_mod)]),
guide = guide_legend(
override.aes = list(
color = rawGeneAnnotationsColors[unique(annotations_filt$product_mod)],
fill = rawGeneAnnotationsColors[unique(annotations_filt$product_mod)],
alpha = 1,
shape = 22,
size = 5
),
nrow = 7,
order = 1
)
) +
scale_color_manual(
"Genomic\nElements",
values = geneAnnotationsColors,
breaks = names(genomicElementsColors),
labels = names(genomicElementsColors),
guide = guide_legend(
override.aes = list(
color = genomicElementsColors,
fill = genomicElementsColors,
alpha = 1,
shape = 22,
size = 5
),
ncol = 1,
order = 2
)
) +
labs(title = "Annotations of SD01 chr11 and chr13", fill = "") +
theme(legend.text = element_text(size = 30), axis.text.x = element_text(size = 30, color = "black"),
plot.title = element_text(size = 30, color = "black"),
strip.text.y = element_text(size = 30, color = "black"),
legend.title = element_text(size = 30, face = "bold"),
legend.box="vertical", legend.margin=margin(),
legend.background = element_blank(),
legend.box.background = element_rect(colour = "black")) ggplotly(sd01_chr11_chr13_annotation_plots)print(sd01_chr11_chr13_annotation_plots)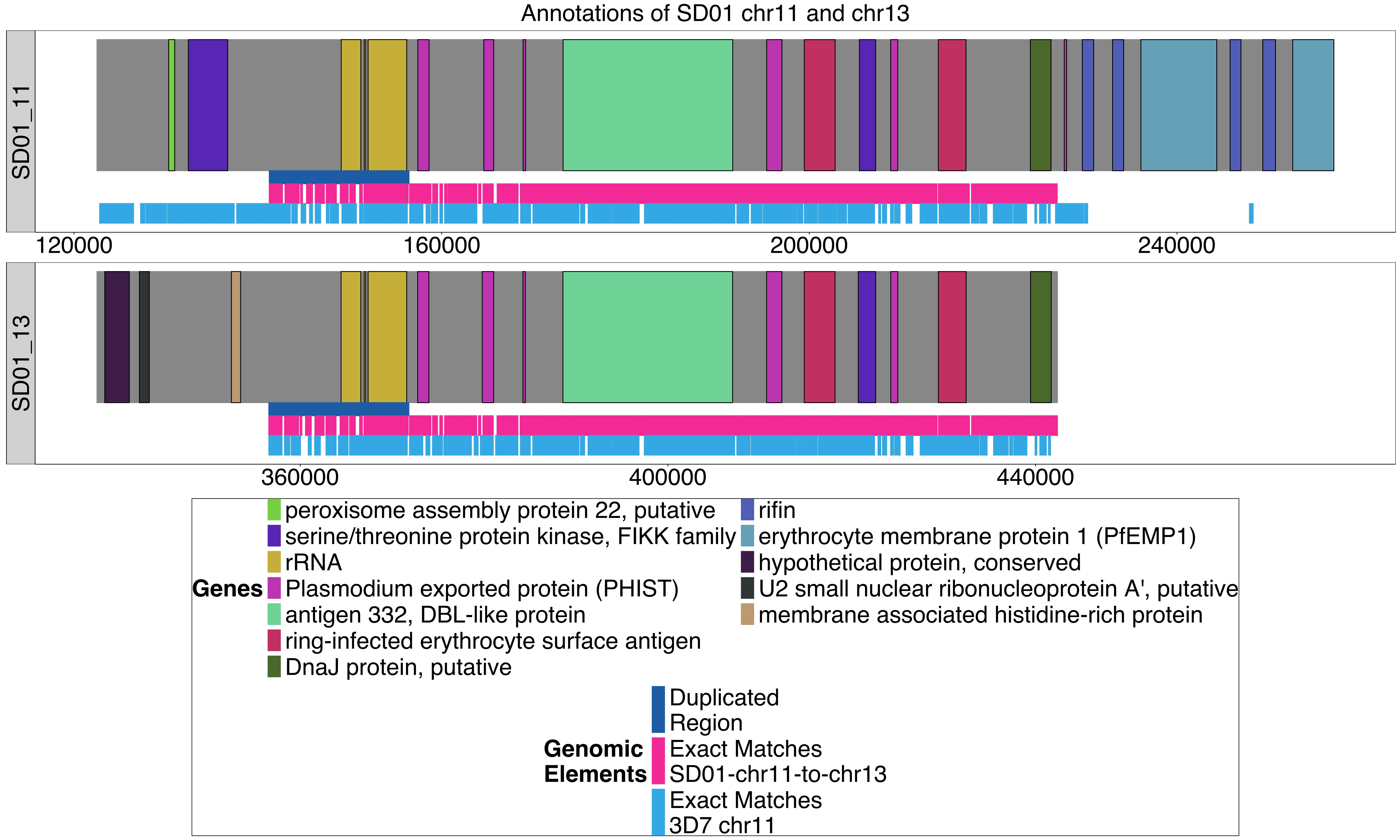
Using nucmer from mummer package(Kurtz et al. 2004)
nucmer /tank/data/genomes/plasmodium//genomes/pf/genomes/Pf3D7.fasta pilon.fasta --prefix PfSD01nano_to_Pf3D7_nucmer
show-coords -T -l -c -H PfSD01nano_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out PfSD01nano_to_Pf3D7_nucmer.delta.bed
elucidator splitColumnContainingMeta --file PfSD01nano_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out PfSD01nano_to_Pf3D7_nucmer.delta.tsv
nucmer /tank/data/genomes/plasmodium//genomes/pf_plusPfSD01/genomes/PfSD01.fasta pilon.fasta --prefix PfSD01nano_to_PfSD01_nucmer
show-coords -T -l -c -H PfSD01nano_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out PfSD01nano_to_PfSD01_nucmer.delta.bed
elucidator splitColumnContainingMeta --file PfSD01nano_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out PfSD01nano_to_PfSD01_nucmer.delta.tsv