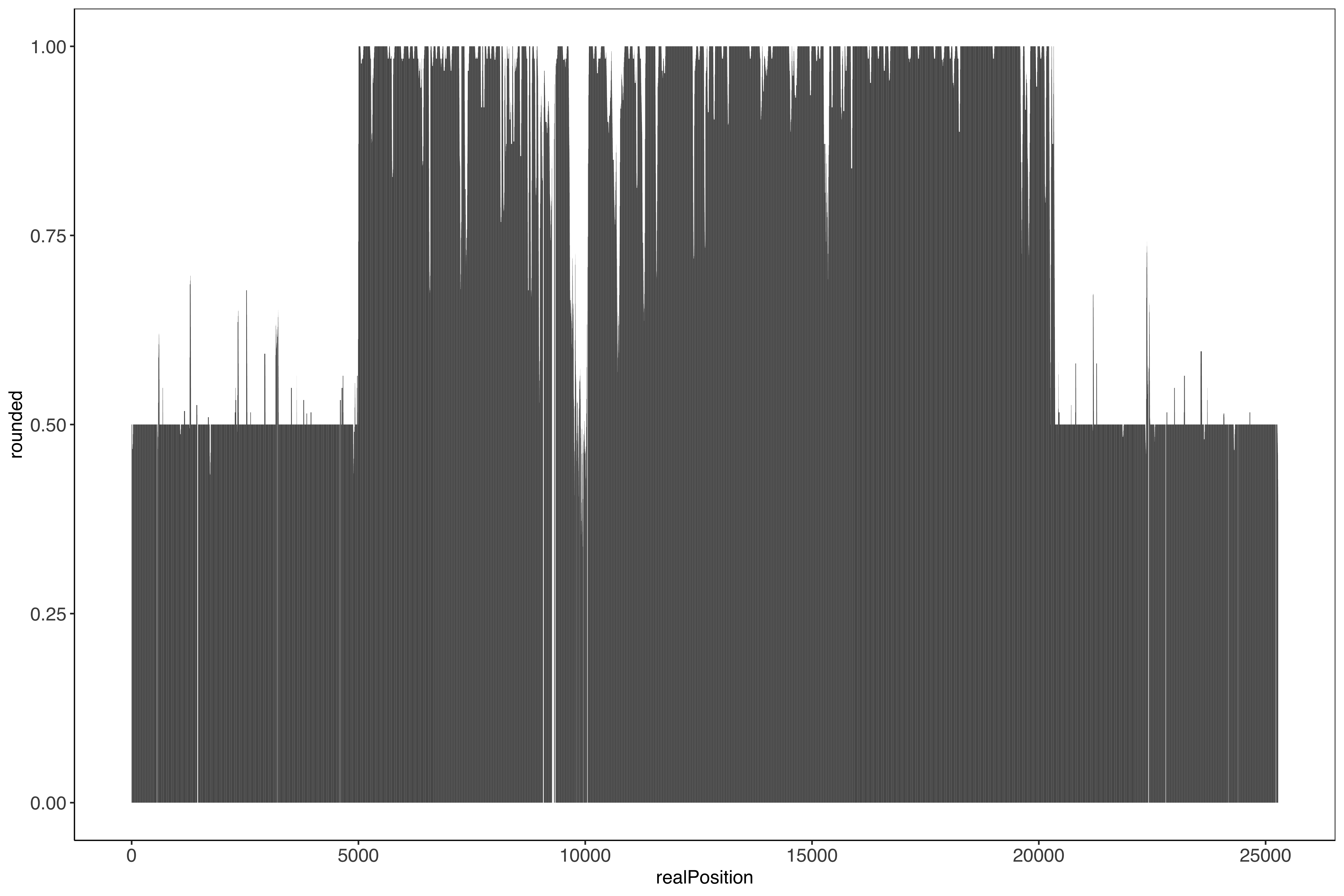
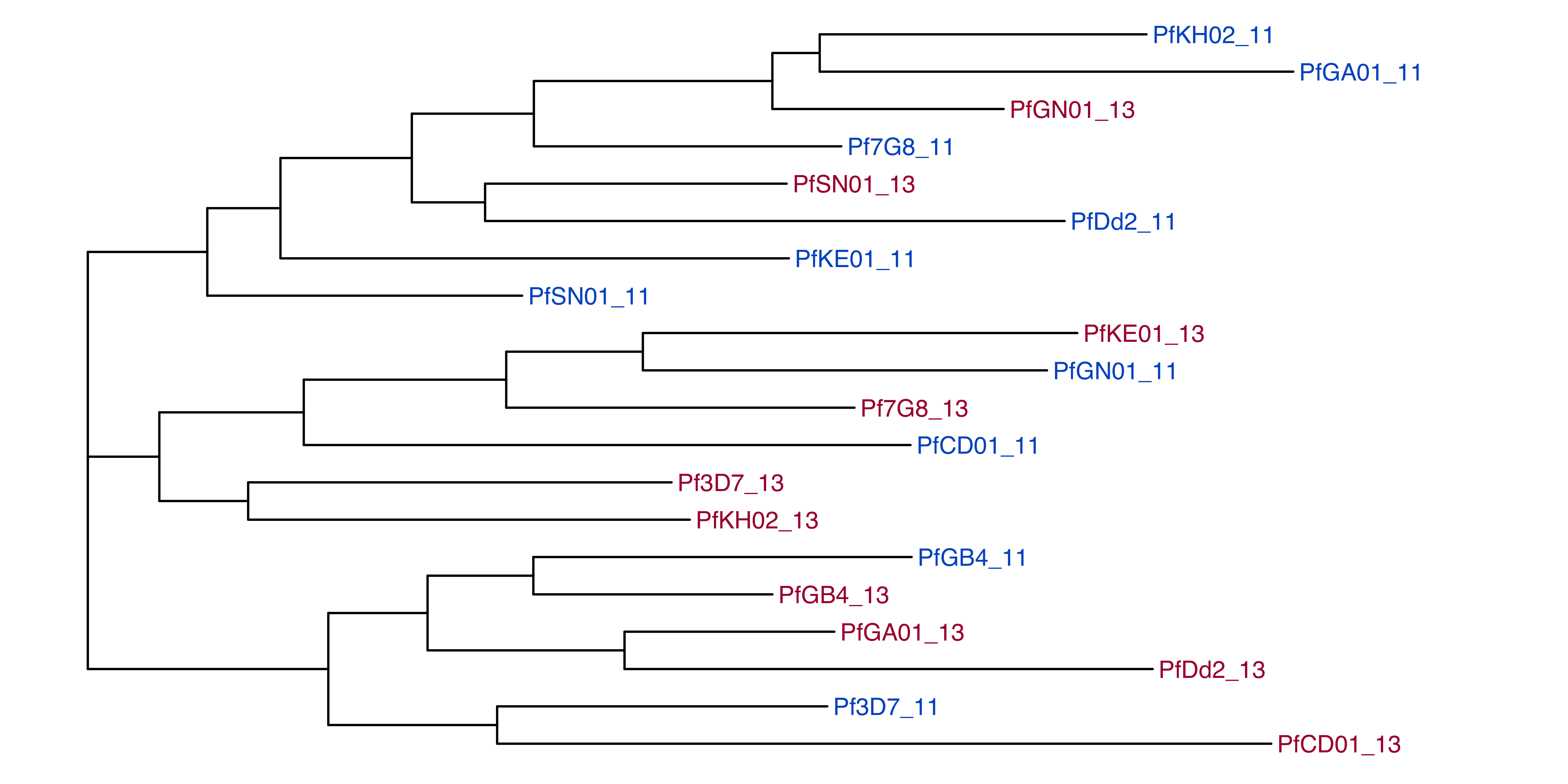
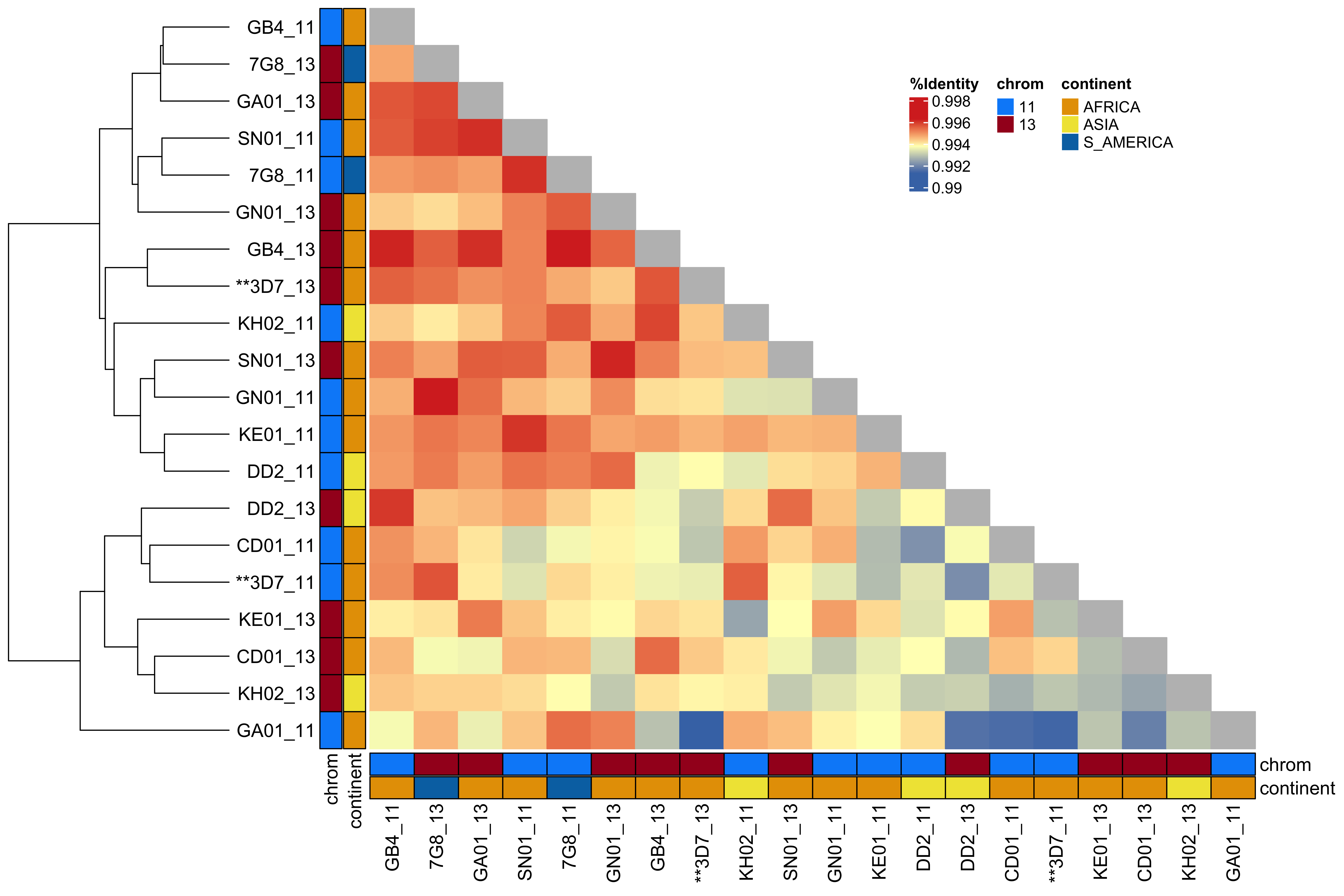
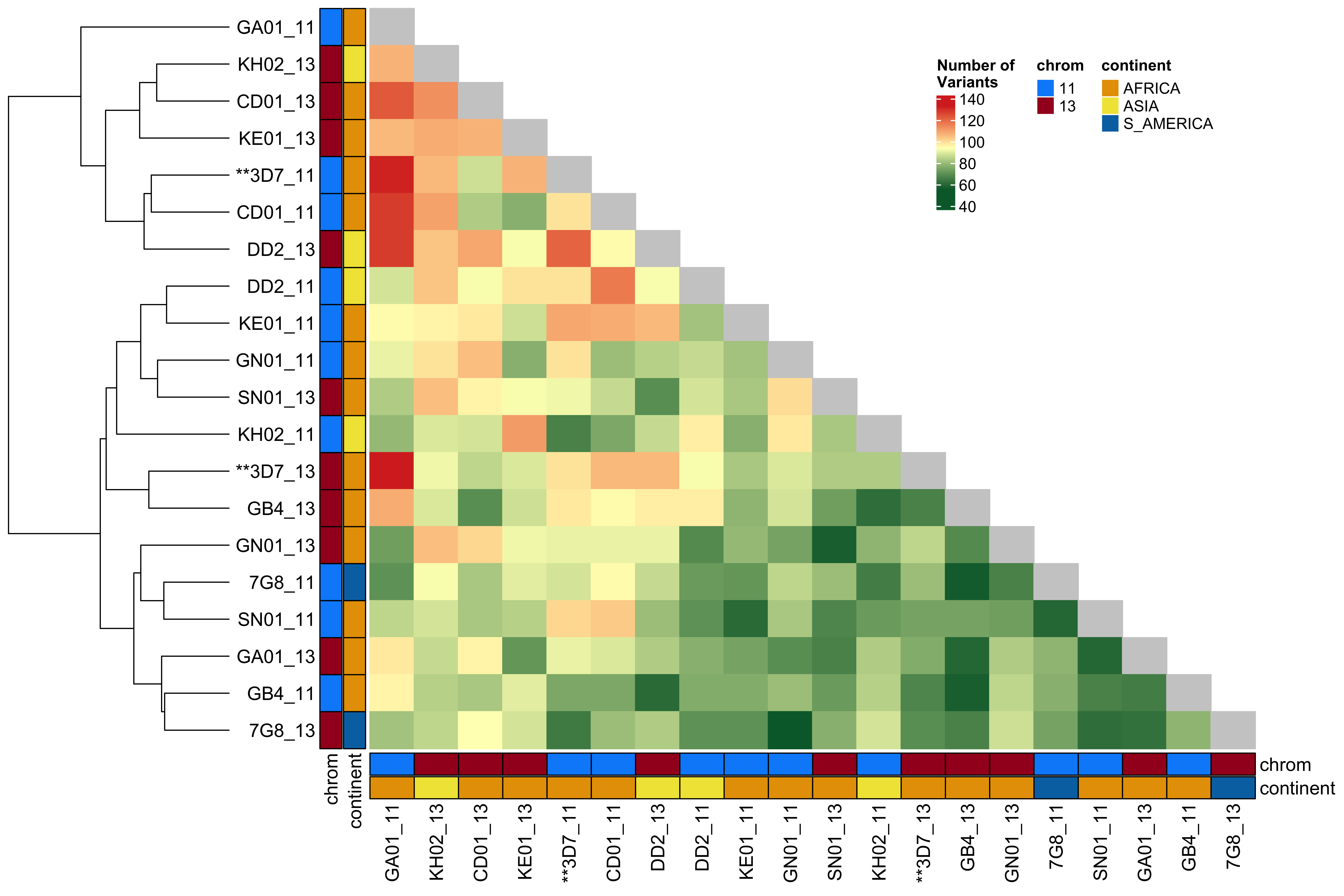
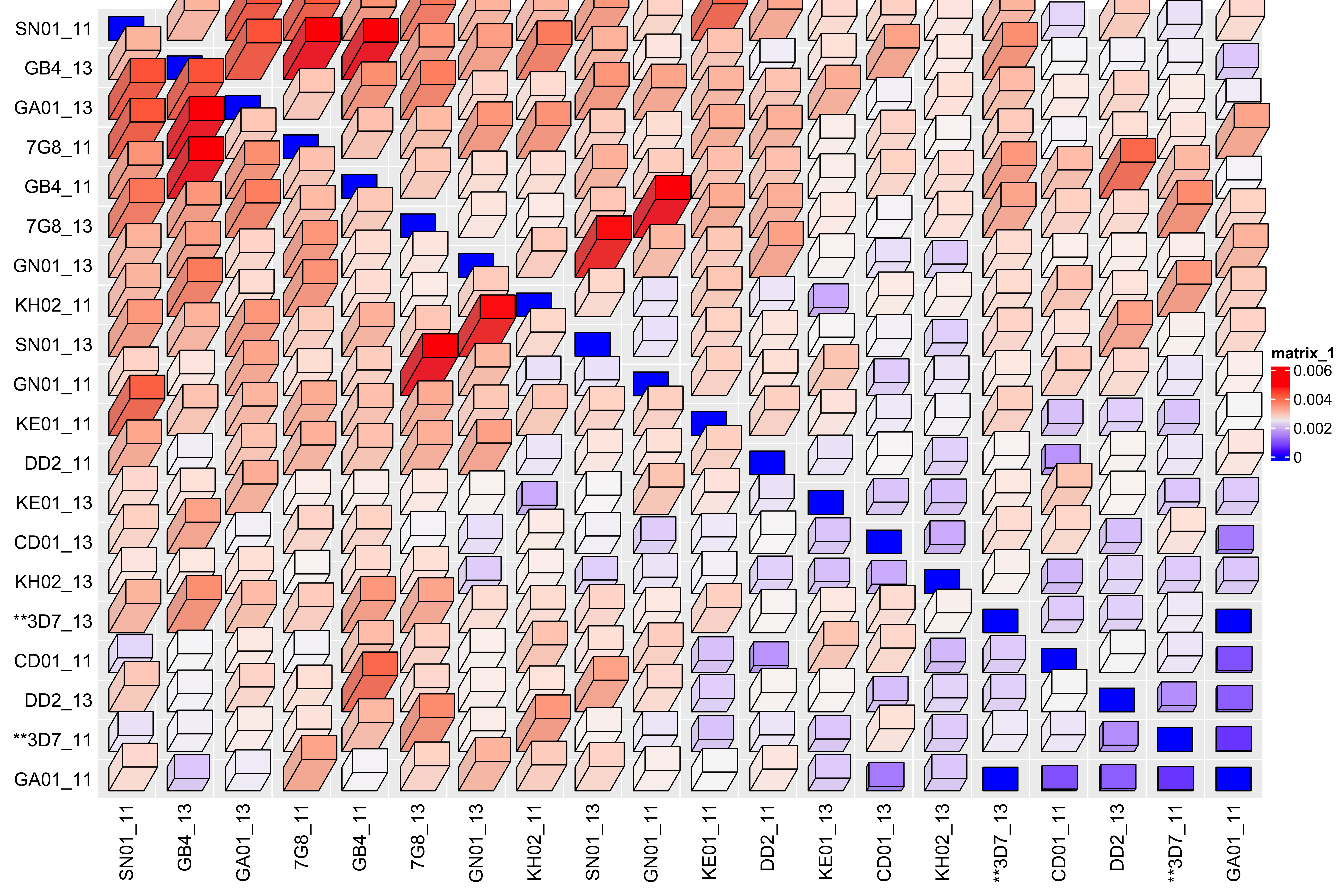
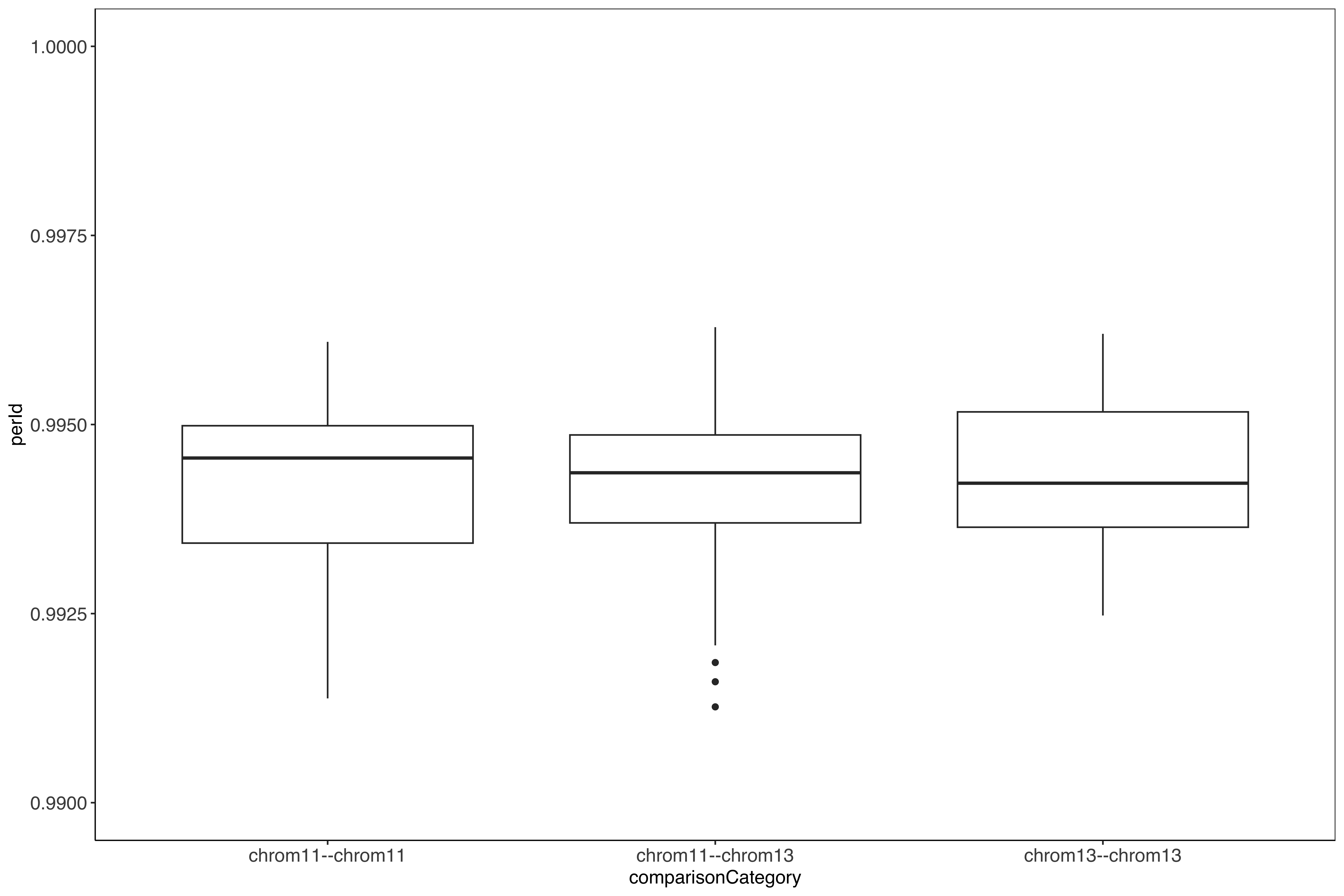
--- title: "Characterizing Duplicated Region" --- ```{r setup, echo=FALSE, message=FALSE} source("../../common.R") ``` ## Investigating shared region within pacbio assemblies[@Otto2018-bb] ```{bash, eval = F} for x in /tank/data/genomes/plasmodium/genomes/pf/genomes/*.fasta; do elucidator splitFile --fasta ${x} --overWrite --trimAtWhiteSpace ; done; ``` ```{r} = c ("Pf3D7" ,"Pf7G8" ,"PfCD01" ,"PfDd2" ,"PfGA01" ,"PfGB4" ,"PfGN01" ,"PfKE01" ,"PfKH02" ,"PfSN01" )cat (strains, sep = " \n " , file = "stableStrains.txt" )= tibble (strain = strains)= strainsDf %>% mutate (chrom11 = paste0 (strain, "_11*.fasta" )) %>% mutate (chrom13 = paste0 (strain, "_13*.fasta" ))= paste0 ("mv " , paste0 (strainsDf$ chrom11, collapse = " " ), " " , paste0 (strainsDf$ chrom13, collapse = " " ), " combinedChrom11_13/" )cat (mvCmd)``` ```{bash, eval = F} elucidator profileSharedKmerBlocks --seq Pf3D7_11_v3.fasta --dout chroms11_13_klen31 --kLen 31 --seqsDir ./ --fasta Pf3D7_11_v3.fasta --overWriteDir cd chroms11_13_klen31 elucidator bedCoordSort --bed perfect.bed | elucidator extendBedRegions --left 5 --right 5 --bed STDIN | bedtools merge | elucidator bed3ToBed6 --bed STDIN | elucidator filterBedRecordsByLength --bed STDIN --minLen 65 | elucidator bedGetIntersectingGenesInGff --gff /work/nhathawa/genomes/pfGenomes_updated/info//gff/Pf3D7.gff --extraAttributes description --overWrite --bed STDIN --out perfectlyShared_minlen65.bed elucidator getFastaWithBed --twoBit /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.2bit --overWrite --bed perfectlyShared_minlen65.bed --out perfectlyShared_minlen65 elucidator extractRefSeqsFromGenomes --genomeDir /tank/data/genomes/plasmodium/genomes/pf/genomes/ --gffDir /tank/data/genomes/plasmodium/genomes/pf/info/gff/ --primaryGenome Pf3D7 --bed perfectlyShared_minlen65.bed --outputDir extractedRefSeqs --identity 95 --coverage 95 --numThreads 15 ``` ```{r} = readr:: read_tsv ("combinedChrom11_13/chroms11_13_klen31/perfectlyShared_minlen65.bed" , col_names = F)= perfectlyShared_minlen65_regions %>% mutate (X4 = paste0 (X4, "-for" ))= tibble ()for (region in perfectlyShared_minlen65_regions$ X4){for (strain in strains){= paste0 ("combinedChrom11_13/chroms11_13_klen31/extractedRefSeqs/" , region, "/beds/" , strain, "_region.bed" )if (file.exists (fnp)){= readr:: read_tsv (fnp, col_names = F, show_col_types = F)$ X4 = region$ strain = strain= bind_rows (= allRegions %>% group_by (strain, X1) %>% summarise (start = min (X2), end = max (X3)) %>% filter (grepl ("_1[13]" , X1)) %>% rename (` #chrom ` = X1) %>% mutate (start = start - 5000 ) %>% mutate (end = end + 5000 ) %>% mutate (name = paste0 (` #chrom ` , "-" , start, "-" , end), len = end - start, strand = "+" ) for (strainName in strains){= allRegions_sum %>% filter (strain == strainName)= allRegions_sum_byStrain %>% group_by () %>% select (- strain)write_tsv (allRegions_sum_byStrain, paste0 ("combinedChrom11_13/chroms11_13_regions/" , strainName, ".bed" ))= allRegions %>% group_by (strain, X1) %>% summarise (start = min (X2), end = max (X3)) %>% filter (grepl ("_1[13]" , X1)) %>% rename (` #chrom ` = X1) %>% mutate (start = start) %>% mutate (end = end) %>% mutate (name = paste0 (` #chrom ` , "-" , start, "-" , end), len = end - start, strand = "+" ) for (strainName in strains){= allRegions_sum_exact %>% filter (strain == strainName)= allRegions_sum_byStrain %>% group_by () %>% select (- strain)write_tsv (allRegions_sum_byStrain, paste0 ("combinedChrom11_13/chroms11_13_regions_exact/" , strainName, ".bed" ))``` ```{bash, eval = F} cd chroms11_13_regions_exact for x in *.bed; do elucidator getFastaWithBed --bed $x --twoBit /tank/data/genomes/plasmodium/genomes/pf/genomes/${x%%.bed}.2bit --overWrite --out ${x%%.bed}.fasta ; done; mkdir sequences cd sequences for x in ../*.fasta; do elucidator splitFile --fasta $x ; done; elucidator profileSharedKmerBlocks --seq Pf3D7_11_v3-1913023-1938396.fasta --dout chroms11_13_klen31 --kLen 31 --seqsDir ./ --fasta Pf3D7_11_v3-1913023-1938396.fasta --overWriteDir ``` ```{bash, eval = F} cd chroms11_13_regions mkdir allSequences for x in Pf*.bed; do elucidator getFastaWithBed --bed $x --twoBit /tank/data/genomes/plasmodium/genomes/pf/genomes/${x%%.bed}.2bit --overWrite --out ${x%%.bed}.fasta ; done; cat *.fasta > allSequences/all.fasta cat Pf*.bed > all.bed cd allSequences muscle -in all.fasta -out aln_all.fasta elucidator trimToLen --fasta aln_all.fasta --length 21945 --overWrite --out trimmed_to_21945_aln_all.fasta elucidator trimFront --fasta trimmed_to_21945_aln_all.fasta --forwardBases 21845 --overWrite --out trimmedFrom_21845_trimmed_to_21945_aln_all.fasta elucidator sortReads --fasta trimmedFrom_21845_trimmed_to_21945_aln_all.fasta --removeGaps --overWrite elucidator determineRegionLastz --individual --fasta sorted_trimmedFrom_21845_trimmed_to_21945_aln_all.fasta --genome /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.2bit -out sorted_trimmedFrom_21845_trimmed_to_21945_aln_all.bed elucidator trimToLen --fasta aln_all.fasta --length 6051 --overWrite --out trimmed_to_6051_aln_all.fasta elucidator trimFront --fasta trimmed_to_6051_aln_all.fasta --forwardBases 5951 --overWrite --out trimmedFrom_5951_trimmed_to_6051_aln_all.fasta elucidator sortReads --fasta trimmedFrom_5951_trimmed_to_6051_aln_all.fasta --removeGaps --overWrite elucidator determineRegionLastz --individual --fasta sorted_trimmedFrom_5951_trimmed_to_6051_aln_all.fasta --genome /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.2bit --overWrite --out sorted_trimmedFrom_5951_trimmed_to_6051_aln_all.bed elucidatorlab getMalnConservedness --fasta aln_all.fasta --blockSize 10 --overWrite --out aln_all_conserved.tab.txt elucidatorlab getAlnPosToRealPosTable --fasta aln_all.fasta --overWrite --out aln_all_position_table.tab.txt ``` ## Beginning of the section  {width=70% fig-align="center"}## Middle  {width=70% fig-align="center"}## End of the section  {width=70% fig-align="center"}## Conserved score ```{r} = readr:: read_tsv ("combinedChrom11_13/chroms11_13_regions/allSequences/aln_all_position_table.tab.txt" ) %>% filter ("Pf3D7_11_v3-1913023-1938396" == name) %>% rename (pos = alignPosition)= readr:: read_tsv ("combinedChrom11_13/chroms11_13_regions/allSequences/aln_all_conserved.tab.txt" )= conservedMulti %>% group_by (pos) %>% filter (frac == max (frac)) %>% filter ("----------" != block ) %>% left_join (conservedPosTab) %>% filter (! is.na (name)) %>% filter ()= 30 = tibble (rawPos = 1 : (nrow (conservedMulti_max) - blockSize),realPosition = numeric (nrow (conservedMulti_max) - blockSize), rounded = numeric (nrow (conservedMulti_max) - blockSize))for (row in 1 : (nrow (conservedMulti_max) - blockSize)){= mean (conservedMulti_max$ frac[row: (row+ blockSize)])if (round > 1 ){print (row)print (blockSize)print (conservedMulti_max$ frac[row: (row+ blockSize)])print (round)stop ()= min (conservedMulti_max$ realPosition[row: (row+ blockSize)])$ realPosition[row] = minPos$ rounded[row] = round= conservedMulti_max_rounding %>% group_by (realPosition) %>% mutate (id = row_number ()) %>% filter (id == 1 ) %>% group_by ()``` ```{r} #| fig-column: screen-inset-shaded ggplot (conservedMulti_max_rounding_filt) + geom_bar (aes (x = realPosition, y = rounded), stat = "identity" )+ ``` ```{bash, eval = F} cd chroms11_13_regions_exact cat *.fasta > all.fasta elucidator compareAllByAll --fasta all.fasta --verbose --numThreads 12 elucidator createSharedSubSegmentsFromRefSeqs --fasta out.fasta --minimumKlen 12 --dout sharedSubSeqs_occurence6 --filterRegionsWithinRegions --refSeqName Pf3D7_11_v3-1918023-1933396 --overWriteDir --genome /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta --minSubRegionLen 150 --maxSubRegionLen 300 --minBlockRegionLen 30 --doNotCollapseLowFreqNodes --correctionOccurenceCutOff 6 --includeFromSubRegionSize 30 muscle -in all.fasta -out aln_all.fasta FastTree -nt aln_all.fasta > aln_all.nwk elucidatorlab getMalnConservedness --fasta aln_all.fasta --blockSize 10 --overWrite --out aln_all_conserved.tab.txt elucidatorlab getAlnPosToRealPosTable --fasta aln_all.fasta --overWrite --out aln_all_position_table.tab.txt elucidator multipleAlnProteinToPcaInput --fasta aln_all.fasta --overWrite --out pcaMat_aln_all.fasta elucidator runTRF --fasta all.fasta --overWriteDir --dout all_trfOutput --supplement --genomicLocation ../all.bed elucidator bedFilterRegionsCompletelyInOther --bed all_trfOutput/genomic_combined.bed --intersectWithBed all_trfOutput/genomic_combined.bed | elucidator bedAddSmartIDForPlotting --bed STDIN --out all_trfOutput/filtered_genomic_combined.bed --overWrite elucidator countBasesPerPosition --fasta aln_all.fasta --overWrite --out aln_all_baseCounts.tab.txt #elucidator createSharedSubSegmentsFromRefSeqs --fasta out.fasta --minimumKlen 12 --dout sharedSubSeqs_occurence6 --filterRegionsWithinRegions --refSeqName Pf3D7_11_v3-1918023-1933396 --overWriteDir --genome /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta --minSubRegionLen 150 --maxSubRegionLen 300 --minBlockRegionLen 30 --doNotCollapseLowFreqNodes --correctionOccurenceCutOff 6 --includeFromSubRegionSize 30 #cd sharedSubSeqs_occurence6/sharedLocs #elucidator bedGetIntersectingGenesInGff --gff /tank/data/genomes/plasmodium/genomes/pf/info/gff/Pf3D7.gff --extraAttributes description --overWrite --bed 0_ref_sharedLocs_genomic.bed --out 0_ref_sharedLocs_genomic_withGeneInfo.bed ``` ## Comparing chromsome 11 and 13 segments ```{r} library (ggtree)= ape:: read.tree ("combinedChrom11_13/chroms11_13_regions_exact/sequences/aln_all.nwk" )$ tip.label = gsub ("_v3" , "" , gsub ("-.*" , "" , tree$ tip.label))= tibble (seq = tree$ tip.label) %>% mutate (chrom = ifelse (grepl ("_11" , seq), "chrom11" , "chrom13" ))= tipDf %>% filter (chrom == "chrom11" )= tipDf %>% filter (chrom == "chrom13" )= list (chrom11 = tipDf_chrom11$ seq, chrom13 = tipDf_chrom13$ seq)= groupOTU (tree, groups)= ggtree:: ggtree (tree, show.legend = FALSE ) + hexpand (.1 ) + geom_tiplab (aes (color = group), show.legend = FALSE ) + scale_color_manual (values = c ("#005AC8" , "#AA0A3C" )) ``` ```{r} #| fig-column: screen-inset-shaded #| fig-height: 5 #| fig-width: 10 print (sharedRegion_pacbioAssembliesPlot)``` ```{r} pdf ("sharedRegion_pacbioAssemblies.pdf" , width = 7.5 , height = 7.5 / 2 , useDingbats = F)print (sharedRegion_pacbioAssembliesPlot)dev.off ()``` ### Comparing Percent Identity ```{r} = readr:: read_tsv ("combinedChrom11_13/chroms11_13_regions_exact/sequences/out.tab.txt" )``` ```{r, eval=FALSE, echo=FALSE} #| fig-column: screen-inset-shaded ggplot(allByAllComp) + geom_tile(aes(x = OtherReadId , y = ReadId, fill = perId)) + geom_text(aes(x = OtherReadId , y = ReadId, label = round(perId*100,2))) + sofonias_theme_xRotate + scale_y_discrete(position = "right") + scale_fill_gradient2(limits = c(min(allByAllComp$perId),1), midpoint = min(allByAllComp$perId) + (max(allByAllComp$perId) - min(allByAllComp$perId))/2 ) ``` ```{r} = allByAllComp %>% select (OtherReadId, ReadId, perId) %>% spread (ReadId, perId)= matrix (c (colnames (allByAllComp_select)[2 ], rep (NA , ncol (allByAllComp_select) - 1 )), nrow = 1 )colnames (addRow) = colnames (allByAllComp_select)= as_tibble (addRow) for (col in 2 : ncol (addRow)){= as.numeric (addRow[,col])= as_tibble (addRow) %>% bind_rows (allByAllComp_select)= as.matrix (allByAllComp_select[,2 : ncol (allByAllComp_select)])rownames (allByAllComp_select_mat) = allByAllComp_select$ OtherReadId= as.matrix (as.dist (allByAllComp_select_mat))0 == allByAllComp_select_mat] = NA ``` ```{r} library (circlize)= readr:: read_tsv ("../../meta/metadata/meta.tab.txt" ) %>% mutate (country = gsub ("South East Asia - East" , "Cambodia" , country))= readr:: read_tsv ("../../meta/metadata/metaByBioSample.tab.txt" ) %>% mutate (country = gsub ("South East Asia - East" , "Cambodia" , country))= allByAllComp_select_mat# rownames(allByAllComp_select_mat_noLabels) = NULL # colnames(allByAllComp_select_mat_noLabels) = NULL rownames (allByAllComp_select_mat_noLabels) = gsub ("Dd2" , "DD2" , gsub ("Pf" , "" , gsub ("_v3" , "" , gsub ("-.*" , "" , rownames (allByAllComp_select_mat_noLabels)))))colnames (allByAllComp_select_mat_noLabels) = gsub ("Dd2" , "DD2" , gsub ("Pf" , "" , gsub ("_v3" , "" , gsub ("-.*" , "" , colnames (allByAllComp_select_mat_noLabels)))))= colorRamp2 (c (min (allByAllComp_select_mat_noLabels, na.rm = T),min (allByAllComp_select_mat_noLabels, na.rm = T) + (max (allByAllComp_select_mat_noLabels, na.rm = T) - min (allByAllComp_select_mat_noLabels, na.rm = T)/ 2 ,max (allByAllComp_select_mat_noLabels, na.rm = T)c ("#4575b4" , "#ffffbf" , "#d73027" ))#c("#ffeda0", "#feb24c", "#f03b20")) = tibble (name = colnames (allByAllComp_select_mat)) %>% mutate (sampchrom = gsub ("_v3" , "" , gsub ("-.*" , "" , name))) %>% mutate (sample = gsub ("Dd2" , "DD2" , gsub ("_.*" , "" , gsub ("Pf" , "" , sampchrom)))) %>% mutate (chrom = gsub (".*_" , "" , sampchrom)) %>% left_join (metaByBioSample %>% select (sample, secondaryRegion))= topAnnoDat[,c ("chrom" , "secondaryRegion" )] %>% rename (continent = secondaryRegion) %>% mutate (continent = gsub ("Netherlands" , "AFRICA" , continent) ) %>% as.data.frame ()= createColorListFromDf (topAnno_df)"continent" ]] = c ("AFRICA" = "#E69F00" , "ASIA" = "#F0E442" , "OCEANIA" = "#F748A5" , "S_AMERICA" = "#0072B2" , "Netherlands" = "black" )"chrom" ]] = c ("11" = "#008DF9" , "13" = "#A40122" )= HeatmapAnnotation (df = topAnno_df,col = topAnnoColors,show_legend = F,gp = gpar (col = "black" )# the annotation direction doesn't appear to work right now # annotation_legend_param = list( # chrom = list(legend_direction = "horizontal", direction = "horizontal"), # continent = list(legend_direction = "horizontal", direction = "horizontal") # ) = rowAnnotation (df = topAnno_df,col = topAnnoColors,show_legend = F,gp = gpar (col = "black" )# , # annotation_legend_param = list( # chrom = list(legend_direction = "horizontal", direction = "horizontal"), # continent = list(legend_direction = "horizontal", direction = "horizontal") # ) colnames (allByAllComp_select_mat_noLabels) = gsub ("3D7" , "**3D7" , colnames (allByAllComp_select_mat_noLabels))rownames (allByAllComp_select_mat_noLabels) = gsub ("3D7" , "**3D7" , rownames (allByAllComp_select_mat_noLabels))= hclust (dist (allByAllComp_select_mat_noLabels))= Heatmap (rect_gp = gpar (type = "none" ),column_dend_side = "bottom" ,cell_fun = function (j, i, x, y, w, h, fill) {if (as.numeric (x) <= 1 - as.numeric (y) + 1e-6 ) {grid.rect (x, y, w, h, gp = gpar (fill = fill, col = fill))# cluster_rows = hc, # cluster_columns = hc, row_names_side = "left" ,name = "%Identity" ,col = col_fun,bottom_annotation = topAnno,left_annotation = sideAnno,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ), show_column_dend = F, show_heatmap_legend = F# , # heatmap_legend_param = list( # direction = "horizontal" # ) = Legend (labels = sort (unique (topAnno_df$ chrom)), title = "chrom" , legend_gp = gpar (fill = topAnnoColors[["chrom" ]][sort (unique (topAnno_df$ chrom))]))= Legend (labels = sort (unique (topAnno_df$ continent)), title = "continent" , legend_gp = gpar (fill = topAnnoColors[["continent" ]][sort (unique (topAnno_df$ continent))]))= Legend (col_fun = col_fun, title = "%Identity" )``` ```{r} #| fig-column: screen-inset-shaded draw (allByAllComp_select_mat_heatmap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "bottom" , annotation_legend_side = "left" )draw (packLegend (heatmap_lgd,chrom_lgd,continent_lgd, direction = "horizontal" ), x = unit (0.775 , "npc" ), y = unit (0.85 , "npc" ))``` ```{r} pdf ("sharedRegion_pacbioAssemblies_percentIdentity.pdf" , width = 7.5 , height = 5 , useDingbats = F)draw (allByAllComp_select_mat_heatmap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "bottom" , annotation_legend_side = "left" )draw (packLegend (heatmap_lgd,chrom_lgd,continent_lgd, direction = "horizontal" ), x = unit (0.775 , "npc" ), y = unit (0.85 , "npc" ))dev.off ()``` ### Comparing Number of variants ```{r} = allByAllComp %>% select (OtherReadId, ReadId, totalDiffs) %>% spread (ReadId, totalDiffs)= matrix (c (colnames (allByAllCompVarCount_select)[2 ], rep (NA , ncol (allByAllCompVarCount_select) - 1 )), nrow = 1 )colnames (addRow) = colnames (allByAllCompVarCount_select)= as_tibble (addRow) for (col in 2 : ncol (addRow)){= as.numeric (addRow[,col])= as_tibble (addRow) %>% bind_rows (allByAllCompVarCount_select)= as.matrix (allByAllCompVarCount_select[,2 : ncol (allByAllCompVarCount_select)])rownames (allByAllCompVarCount_select_mat) = allByAllCompVarCount_select$ OtherReadId= as.matrix (as.dist (allByAllCompVarCount_select_mat))0 == allByAllCompVarCount_select_mat] = NA ``` ```{r} library (circlize)= readr:: read_tsv ("../../meta/metadata//meta.tab.txt" ) %>% mutate (country = gsub ("South East Asia - East" , "Cambodia" , country))= readr:: read_tsv ("../../meta/metadata/metaByBioSample.tab.txt" ) %>% mutate (country = gsub ("South East Asia - East" , "Cambodia" , country))= allByAllCompVarCount_select_mat# rownames(allByAllCompVarCount_select_mat_noLabels) = NULL # colnames(allByAllCompVarCount_select_mat_noLabels) = NULL rownames (allByAllCompVarCount_select_mat_noLabels) = gsub ("Dd2" , "DD2" , gsub ("Pf" , "" , gsub ("_v3" , "" , gsub ("-.*" , "" , rownames (allByAllCompVarCount_select_mat_noLabels)))))colnames (allByAllCompVarCount_select_mat_noLabels) = gsub ("Dd2" , "DD2" , gsub ("Pf" , "" , gsub ("_v3" , "" , gsub ("-.*" , "" , colnames (allByAllCompVarCount_select_mat_noLabels)))))= colorRamp2 (c (min (allByAllCompVarCount_select_mat_noLabels, na.rm = T),min (allByAllCompVarCount_select_mat_noLabels, na.rm = T) + (max (allByAllCompVarCount_select_mat_noLabels, na.rm = T) - min (allByAllCompVarCount_select_mat_noLabels, na.rm = T)/ 2 ,max (allByAllCompVarCount_select_mat_noLabels, na.rm = T)c ("#006837" , "#ffffbf" , "#d73027" ))#c("#4575b4", "#ffffbf", "#d73027")) #c("#ffeda0", "#feb24c", "#f03b20")) = tibble (name = colnames (allByAllCompVarCount_select_mat)) %>% mutate (sampchrom = gsub ("_v3" , "" , gsub ("-.*" , "" , name))) %>% mutate (sample = gsub ("Dd2" , "DD2" , gsub ("_.*" , "" , gsub ("Pf" , "" , sampchrom)))) %>% mutate (chrom = gsub (".*_" , "" , sampchrom)) %>% left_join (metaByBioSample %>% select (sample, secondaryRegion))= topAnnoDat[,c ("chrom" , "secondaryRegion" )] %>% rename (continent = secondaryRegion) %>% mutate (continent = gsub ("Netherlands" , "AFRICA" , continent) ) %>% as.data.frame ()= createColorListFromDf (topAnno_df)"continent" ]] = c ("AFRICA" = "#E69F00" , "ASIA" = "#F0E442" , "OCEANIA" = "#F748A5" , "S_AMERICA" = "#0072B2" , "Netherlands" = "black" )"chrom" ]] = c ("11" = "#008DF9" , "13" = "#A40122" )= HeatmapAnnotation (df = topAnno_df,col = topAnnoColors,show_legend = F,gp = gpar (col = "black" )# the annotation direction doesn't appear to work right now # annotation_legend_param = list( # chrom = list(legend_direction = "horizontal", direction = "horizontal"), # continent = list(legend_direction = "horizontal", direction = "horizontal") # ) = rowAnnotation (df = topAnno_df,col = topAnnoColors,show_legend = F,gp = gpar (col = "black" )# , # annotation_legend_param = list( # chrom = list(legend_direction = "horizontal", direction = "horizontal"), # continent = list(legend_direction = "horizontal", direction = "horizontal") # ) colnames (allByAllCompVarCount_select_mat_noLabels) = gsub ("3D7" , "**3D7" , colnames (allByAllCompVarCount_select_mat_noLabels))rownames (allByAllCompVarCount_select_mat_noLabels) = gsub ("3D7" , "**3D7" , rownames (allByAllCompVarCount_select_mat_noLabels))= hclust (dist (allByAllCompVarCount_select_mat_noLabels))= Heatmap (rect_gp = gpar (type = "none" ),column_dend_side = "bottom" ,cell_fun = function (j, i, x, y, w, h, fill) {if (as.numeric (x) <= 1 - as.numeric (y) + 1e-6 ) {grid.rect (x, y, w, h, gp = gpar (fill = fill, col = fill))# cluster_rows = hc, # cluster_columns = hc, row_names_side = "left" ,name = "Number of \n Variants" ,col = col_fun,bottom_annotation = topAnno,left_annotation = sideAnno,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ), show_column_dend = F, show_heatmap_legend = F,na_col = "grey80" ,# , # heatmap_legend_param = list( # direction = "horizontal" # ) = Legend (labels = sort (unique (topAnno_df$ chrom)), title = "chrom" , legend_gp = gpar (fill = topAnnoColors[["chrom" ]][sort (unique (topAnno_df$ chrom))]))= Legend (labels = sort (unique (topAnno_df$ continent)), title = "continent" , legend_gp = gpar (fill = topAnnoColors[["continent" ]][sort (unique (topAnno_df$ continent))]))= Legend (col_fun = col_fun, title = "Number of \n Variants" )``` ```{r} #| fig-column: screen-inset-shaded draw (allByAllCompVarCount_select_mat_heatmap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "bottom" , annotation_legend_side = "left" )draw (packLegend (heatmap_lgd,chrom_lgd,continent_lgd, direction = "horizontal" ), x = unit (0.80 , "npc" ), y = unit (0.85 , "npc" ))``` ```{r} pdf ("sharedRegion_pacbioAssemblies_varCount.pdf" , width = 7.5 , height = 5 , useDingbats = F)draw (allByAllCompVarCount_select_mat_heatmap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "bottom" , annotation_legend_side = "left" )draw (packLegend (heatmap_lgd,chrom_lgd,continent_lgd, direction = "horizontal" ), x = unit (0.80 , "npc" ), y = unit (0.85 , "npc" ))dev.off ()``` ```{r, eval = F, echo=F} library(gridExtra) conservedInfo_between_11_and_13_sharedRegion = readr::read_tsv("../../MappingOutSurroundingRegions/conservedInfo_between_11_and_13_sharedRegion.tsv") conservedInfo_current_plot = ggplot(conservedInfo_between_11_and_13_sharedRegion) + geom_segment( aes( x = chrom1Pos_start, xend = crhom1Pos_end - 1, y = chrom2Pos, yend = chrom2Pos + size - 1 ), linewidth = 2.5, color = "black" ) + geom_rect( ymin = conservedInfo_between_11_and_13_sharedRegion$chrom2_section_start[1], ymax = conservedInfo_between_11_and_13_sharedRegion$chrom2_section_start[1] + conservedInfo_between_11_and_13_sharedRegion$chrom2_section_len[1], xmin = conservedInfo_between_11_and_13_sharedRegion$chrom1_section_start[1] - conservedInfo_between_11_and_13_sharedRegion$chrom1_section_len[1] * 0.1, xmax = conservedInfo_between_11_and_13_sharedRegion$chrom1_section_start[1] - conservedInfo_between_11_and_13_sharedRegion$chrom1_section_len[1] * 0.001, fill = "grey81") + geom_rect( ymax = conservedInfo_between_11_and_13_sharedRegion$chrom2_section_start[1] - conservedInfo_between_11_and_13_sharedRegion$chrom2_section_len[1] * 0.001, ymin = conservedInfo_between_11_and_13_sharedRegion$chrom2_section_start[1] - conservedInfo_between_11_and_13_sharedRegion$chrom2_section_len[1] * 0.1, xmin = conservedInfo_between_11_and_13_sharedRegion$chrom1_section_start[1], xmax = conservedInfo_between_11_and_13_sharedRegion$chrom1_section_start[1] + conservedInfo_between_11_and_13_sharedRegion$chrom1_section_len[1], fill = "grey81") + geom_rect(data = pf3d7Genes_chrom1, aes(xmin = col.1, xmax = col.2, ymax = conservedInfo_between_11_and_13_sharedRegion$chrom2_section_start[1] - conservedInfo_between_11_and_13_sharedRegion$chrom2_section_len[1] * 0.001, ymin = conservedInfo_between_11_and_13_sharedRegion$chrom2_section_start[1] - conservedInfo_between_11_and_13_sharedRegion$chrom2_section_len[1] * 0.1, fill = description)) + geom_rect(data = pf3d7Genes_chrom2, aes(ymin = col.1, ymax = col.2, xmax = conservedInfo_between_11_and_13_sharedRegion$chrom1_section_start[1] - conservedInfo_between_11_and_13_sharedRegion$chrom1_section_len[1] * 0.001, xmin = conservedInfo_between_11_and_13_sharedRegion$chrom1_section_start[1] - conservedInfo_between_11_and_13_sharedRegion$chrom1_section_len[1] * 0.1, fill = description)) + labs(title = "", x = "3D7 Chr13-2792021-2807295 (bp=15,274)", y = "3D7 Chr11-1918028-1933288 (bp=15,260)", fill = "") + sofonias_theme + coord_equal() + # scale_fill_tableau() + scale_fill_manual(values = c("#2271B2", "#F748A5", "#3DB7E9", "#D55E00")) + guides(fill=guide_legend(ncol = 1,byrow=TRUE)) + theme(legend.position = c(0.3, 0.7), panel.border = element_blank(), legend.background = element_blank(), legend.box.background = element_rect(colour = "black")) grobHeatmap = grid.grabExpr({ draw(allByAllComp_select_mat_heatmap, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "left") draw(packLegend(heatmap_lgd,chrom_lgd,continent_lgd, direction = "horizontal"), x = unit(0.775, "npc"), y = unit(0.85, "npc")) }) grobGgplot = grid.grabExpr({ print(conservedInfo_current_plot) }) gl = list(grobGgplot, grobHeatmap) lay <- rbind(c(1,1,1,1,NA,NA,NA,NA,NA,NA,NA,NA,NA,NA), c(1,1,1,1,NA,NA,2,2,2,2,2,2,2,2), c(1,1,1,1,NA,NA,2,2,2,2,2,2,2,2), c(1,1,1,1,NA,NA,2,2,2,2,2,2,2,2), c(1,1,1,1,NA,NA,2,2,2,2,2,2,2,2), c(1,1,1,1,NA,NA,NA,NA,NA,NA,NA,NA,NA,NA)) pdf("test.pdf", width = 10, height = 10, useDingbats = F) grid.arrange( grobs = gl, layout_matrix = lay ) dev.off() ``` ```{r} #| fig-column: screen-inset-shaded = allByAllComp_select_mat_noLabels= allByAllComp_select_mat_noLabels_mod - min (allByAllComp_select_mat_noLabels_mod, na.rm = T)is.na (allByAllComp_select_mat_noLabels_mod)] = 0 Heatmap3D (allByAllComp_select_mat_noLabels_mod - min (allByAllComp_select_mat_noLabels_mod))``` ## Chromosome vs chromosome percent identity [ chr05 and chr07 chromosomes rRNA loci ](../sharedBetween05_and_07/characterizingSharingInPacbio.qmd#chromosome-vs-chromosome-percent-identity) where the same chromosomes are more closely related than between chromosomes. Could be suggestive that chr11 and chr13 are inter-combining more than chr05 and chr07 ```{r} = allByAllComp %>% group_by (ReadId, OtherReadId) %>% mutate (read1_chrom = ifelse (grepl ("_11" , ReadId), "chrom11" , "chrom13" ))%>% mutate (read2_chrom = ifelse (grepl ("_11" , OtherReadId), "chrom11" , "chrom13" )) %>% mutate (comparisonCategory = paste0 (sort (c (read1_chrom, read2_chrom)), collapse = "--" ) ) %>% ungroup ()ggplot (allByAllComp_mod) + geom_boxplot (aes (x = comparisonCategory, y = perId)) + + #scale_y_continuous(limits = c(min(allByAllComp_mod$perId), 1)) + scale_y_continuous (limits = c (0.990 , 1 ))``` # Writing out region ```{r} = tibble (` #chrom ` = c ("Pf3D7_11_v3" , "Pf3D7_13_v3" ), start = c (1918029 , 2792022 ), end = c (1933390 , 2807397 ), name = c ("Pf3D7_11_v3-1918029-1918124--Pf3D7_11_v3-1933291-1933390__Pf3D7_13_v3-2792022-2792117--Pf3D7_13_v3-2807298-2807397" , "Pf3D7_11_v3-1918029-1918124--Pf3D7_11_v3-1933291-1933390__Pf3D7_13_v3-2792022-2792117--Pf3D7_13_v3-2807298-2807397" )= outBed %>% mutate (len = end - start) %>% mutate (strand = "+" )create_dt (outBed)write_tsv (outBed, "investigatingChrom11Chrom13/shared_11_13_region.bed" )``` <!-- ## Mapping out shared region --> <!-- ```{r} --> <!-- conservedPosTab = readr::read_tsv("combinedChrom11_13/chroms11_13_regions_exact/sequences/aln_all_position_table.tab.txt") %>% --> <!-- separate(name, into = c("chrom", "start", "end"), convert = T, remove = F, sep = "-") %>% --> <!-- mutate(realPosition = realPosition + start) --> <!-- aln_all_baseCounts = readr::read_tsv("combinedChrom11_13/chroms11_13_regions_exact/sequences/aln_all_baseCounts.tab.txt") --> <!-- aln_all_baseCounts_filt = aln_all_baseCounts %>% --> <!-- filter(count > 0, fraction <1) %>% --> <!-- group_by(position) %>% --> <!-- filter(max(count) < 19) %>% --> <!-- filter(!any(base == '-')) %>% --> <!-- select(-meanReadLengthRounded) --> <!-- conservedPosTab_Pf3D7_11_v3 = conservedPosTab %>% --> <!-- filter(grepl("Pf3D7_11_v3", name)) %>% --> <!-- rename(position = alignPosition) %>% --> <!-- left_join(aln_all_baseCounts_filt)%>% --> <!-- filter(!is.na(base)) --> <!-- conservedPosTab_Pf3D7_13_v3 = conservedPosTab %>% --> <!-- filter(grepl("Pf3D7_13_v3", name))%>% --> <!-- rename(position = alignPosition) %>% --> <!-- left_join(aln_all_baseCounts_filt) %>% --> <!-- filter(!is.na(base)) --> <!-- variableSitesInPacbioGenomes = bind_rows( --> <!-- conservedPosTab_Pf3D7_11_v3, --> <!-- conservedPosTab_Pf3D7_13_v3 --> <!-- ) %>% --> <!-- mutate(`#chrom` = chrom) --> <!-- chroms = readr::read_tsv("../../MappingOutSurroundingRegions/surroundingRegionsMaterials/chromLengths/Pf3D7.txt", col_names = c("chrom", "length")) --> <!-- chrom11_genes = readr::read_tsv("../../MappingOutSurroundingRegions/surroundingRegionsMaterials//endBeds/split_Pf3D7_chrom11_toEnd_genes.tab.txt", col_names = T) --> <!-- chrom13_genes = readr::read_tsv("../../MappingOutSurroundingRegions/surroundingRegionsMaterials//endBeds/split_Pf3D7_chrom13_toEnd_genes.tab.txt", col_names = T) --> <!-- allGenes = bind_rows(chrom11_genes, chrom13_genes) %>% --> <!-- rename(chrom = col.0, start = col.1, end = col.2, name = col.3, length = col.4, strand = col.5) %>% --> <!-- mutate(rawGeneDescription = description) %>% --> <!-- mutate(gene = ifelse(grepl("PF3D7_0831800", description), "HRP II", "other")) %>% --> <!-- mutate(gene = ifelse(grepl("PF3D7_1372200", description), "HRP III", gene)) %>% --> <!-- mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>% --> <!-- mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>% --> <!-- mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>% --> <!-- mutate(description = ifelse("membrane associated erythrocyte binding-like protein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>% --> <!-- mutate(description = ifelse("membrane associated erythrocyte binding-likeprotein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>% --> <!-- mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description)) %>% --> <!-- mutate(description = gsub(",putative", ", putative", description)) %>% --> <!-- mutate(description = gsub(",pseudogene", ", pseudogene", description))%>% --> <!-- mutate(description = gsub("unknownfunction", "unknown function", description))%>% --> <!-- mutate(description = gsub("conserved protein, unknown function", "conserved Plasmodium protein, unknown function", description)) %>% --> <!-- mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>% --> <!-- mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>% --> <!-- mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>% --> <!-- mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description))%>% --> <!-- mutate(description = gsub(",pseudogene", ", pseudogene", description))%>% --> <!-- mutate(description = gsub("unknownfunction", "unknown function", description))%>% --> <!-- mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>% --> <!-- mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>% --> <!-- mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>% --> <!-- mutate(description = ifelse(grepl("sporozoite and liver stage tryptophan-rich protein, putative", description), "tryptophan/threonine-rich antigen", description))%>% --> <!-- mutate(description = ifelse(grepl("CRA domain-containing protein, putative", description), "conserved Plasmodium protein, unknown function", description))%>% --> <!-- mutate(description = gsub(",pseudogene", ", pseudogene", description)) %>% --> <!-- mutate(description = gsub("surfaceantigen", "surface antigen", description)) %>% --> <!-- mutate(description = gsub("Tetratricopeptide repeat, putative", "tetratricopeptide repeat protein, putative", description)) %>% --> <!-- mutate(description = gsub("transmembraneprotein", "transmembrane protein", description)) %>% --> <!-- mutate(description = ifelse(grepl("PfEMP1", description) & grepl("pseudogene", description), "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>% --> <!-- mutate(description = gsub("PIR protein", "stevor", description)) %>% --> <!-- mutate(description = gsub("erythrocyte membrane protein 1-like", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>% --> <!-- mutate(description = gsub("acidic terminal segments, variant surface antigen of PfEMP1, putative", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description))%>% --> <!-- mutate(description = ifelse(grepl("CoA binding protein", description, ignore.case = T), "acyl-CoA binding protein", description)) %>% --> <!-- mutate(description = ifelse(grepl("transfer RNA", description) | grepl("tRNA", description), "tRNA", description))%>% --> <!-- mutate(description = ifelse(grepl("cytoadherence", description), "CLAG", description))%>% --> <!-- mutate(description = ifelse(grepl("surface-associated interspersed protein", description), "SURFIN", description))%>% --> <!-- mutate(description = ifelse(grepl("SURFIN", description), "SURFIN", description))%>% --> <!-- mutate(description = ifelse(grepl("stevor-like", description), "stevor, pseudogene", description)) %>% --> <!-- mutate(description = ifelse(grepl("exported protein family", description), "exported protein family", description)) %>% --> <!-- mutate(description = ifelse(grepl("ribosomal RNA", description), "rRNA", description)) %>% --> <!-- mutate(description = ifelse(grepl("serine/threonine protein kinase", description), "serine/threonine protein kinase, FIKK family", description)) %>% --> <!-- mutate(description = ifelse(grepl("hypothetical protein", description), "hypothetical protein, conserved", description)) %>% --> <!-- mutate(description = ifelse(grepl("conserved Plasmodium protein, unknown function", description), "hypothetical protein, conserved", description)) %>% --> <!-- mutate(description = ifelse(grepl("Rifin/stevor family, putative", description), "stevor", description)) %>% --> <!-- mutate(description = ifelse(grepl("stevor", description), "stevor", description)) %>% --> <!-- mutate(description = ifelse(grepl("rifin", description), "rifin", description)) %>% --> <!-- mutate(description = ifelse(grepl("erythrocyte membrane protein 1 (PfEMP1), pseudogene", description), "erythrocyte membrane protein 1 (PfEMP1)", description)) %>% --> <!-- mutate(description = ifelse(grepl("erythrocyte membrane protein 1", description), "erythrocyte membrane protein 1 (PfEMP1)", description)) %>% --> <!-- mutate(description = ifelse(grepl("probably protein", description), "unspecified product", description)) %>% --> <!-- mutate(description = ifelse(grepl("RESA", description), "RESA", description)) %>% --> <!-- mutate(description = ifelse(grepl("ring-infected erythrocyte surface antigen", description), "ring-infected erythrocyte surface antigen", description))%>% --> <!-- mutate(description = ifelse(grepl("Duffy binding domain/Erythrocyte binding antigen175, putative", description), "erythrocyte binding like protein 1", description)) %>% --> <!-- mutate(description = ifelse(grepl("erythrocyte binding like protein 1", description), "erythrocyte binding like protein 1", description))%>% --> <!-- mutate(description = ifelse(grepl("unspecified product", description), "hypothetical protein, conserved", description))%>% --> <!-- mutate(description = ifelse(grepl("probable protein, unknown function", description), "hypothetical protein, conserved", description)) %>% --> <!-- mutate(description = ifelse(grepl("RESA", description), "ring-infected erythrocyte surface antigen", description)) --> <!-- descriptionColorsNames = unique(c(allGenes$description)) --> <!-- descriptionColors = scheme$hex(length(descriptionColorsNames)) --> <!-- names(descriptionColors) = descriptionColorsNames --> <!-- renamed_combined_sharedRegionsNotInTandems = readr::read_tsv("../../sharedBetween11_and_13/windows/renamed_combined_sharedRegionsNotInTandems.bed", col_names = F) --> <!-- combined_11_13_sharedRegions_stepped = readr::read_tsv("../../sharedBetween11_and_13/windows/combined_11_13_sharedRegions_stepped.bed", col_names = F); --> <!-- inbetweenConservedRegions = readr::read_tsv("../../sharedBetween11_and_13/windows/inbetweenConservedRegions.bed", col_names = F); --> <!-- sharedRegionsNotInTandems_size150_step25 = readr::read_tsv("../../sharedBetween11_and_13/windows/sharedRegionsNotInTandems_size150_step25.bed", col_names = F); --> <!-- bedsOfSharedRegions = readr::read_tsv("../../sharedBetween11_and_13/bedsOfSharedRegions/Pf3D7.bed", col_names = T); --> <!-- inbetweenLargeTandems = readr::read_tsv("../../windows/inbetweenLargeTandems.bed", col_names = F); --> <!-- finalWindows_inSharedRegion = readr::read_tsv("../windowAnalysis/windows/finalHRPII_HRPIII_windows.bed", col_names = F) %>% --> <!-- left_join(outBed %>% --> <!-- select(1:3) %>% --> <!-- rename(X1 = `#chrom`, --> <!-- sharedStart = start, --> <!-- sharedEnd = end)) %>% --> <!-- filter((X2 >= sharedStart & X2 < sharedEnd) | (X3 >= sharedStart & X3 <= sharedEnd)) --> <!-- finalSubWindows_inSharedRegion = readr::read_tsv("../windowAnalysis/windows/finalHRPII_HRPIII_windows_combinedVarConservedRegions.bed", col_names = F) %>% --> <!-- left_join(outBed %>% --> <!-- select(1:3) %>% --> <!-- rename(X1 = `#chrom`, --> <!-- sharedStart = start, --> <!-- sharedEnd = end)) %>% --> <!-- filter((X2 >= sharedStart & X2 < sharedEnd) | (X3 >= sharedStart & X3 <= sharedEnd)) --> <!-- allGenes_inSharedRegion = allGenes %>% --> <!-- left_join(outBed %>% --> <!-- select(1:3) %>% --> <!-- rename(chrom = `#chrom`, --> <!-- sharedStart = start, --> <!-- sharedEnd = end)) %>% --> <!-- filter((start >= sharedStart & start < sharedEnd) | (end >= sharedStart & end <= sharedEnd)) %>% --> <!-- group_by(chrom, start, end, name, length) %>% --> <!-- mutate(start = max(start, sharedStart), --> <!-- end = min(end, sharedEnd)) --> <!-- genomic_combined_filtered = readr::read_tsv("chroms11_13_regions_exact/sequences/all_trfOutput/filtered_genomic_combined.bed", col_names = F) --> <!-- genomic_combined_filtered_3d7 = genomic_combined_filtered %>% --> <!-- filter(X1 %in% c("Pf3D7_11_v3", "Pf3D7_13_v3")) --> <!-- colnames(genomic_combined_filtered_3d7)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- colnames(finalWindows_inSharedRegion)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- colnames(finalSubWindows_inSharedRegion)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- colnames(allGenes_inSharedRegion)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- colnames(renamed_combined_sharedRegionsNotInTandems)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- colnames(combined_11_13_sharedRegions_stepped)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- colnames(inbetweenConservedRegions)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- colnames(sharedRegionsNotInTandems_size150_step25)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- colnames(bedsOfSharedRegions)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- colnames(inbetweenLargeTandems)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- inbetweenLargeTandems_inSharedRegion = inbetweenLargeTandems%>% --> <!-- left_join(outBed %>% --> <!-- select(1:3) %>% --> <!-- rename( --> <!-- sharedStart = start, --> <!-- sharedEnd = end)) %>% --> <!-- filter((start >= sharedStart & start < sharedEnd) | (end >= sharedStart & end <= sharedEnd)) %>% --> <!-- group_by(`#chrom`, start, end, name, len) %>% --> <!-- mutate(start = max(start, sharedStart), --> <!-- end = min(end, sharedEnd)) %>% --> <!-- ungroup() --> <!-- # temp = readr::read_tsv("tunning/shared_11_13_region_trfOutput/onlymapped_nochr11Locs_windowsAcrossDuplicatedRegion_chr11_step20_size200_id90.bed", col_names = F) %>% --> <!-- # group_by(X4) %>% --> <!-- # count() %>% --> <!-- # ungroup() %>% --> <!-- # separate(X4, remove = F, convert = T, sep = "-", into = c("chrom", "start", "end", "strand")) %>% --> <!-- # mutate("#chrom" = chrom) --> <!-- # temp = readr::read_tsv("outFilt_id80.tab.txt", col_names = T) %>% --> <!-- # mutate("#chrom" = chrom) --> <!-- # --> <!-- # temp2 = readr::read_tsv("outFilt_id90.tab.txt", col_names = T) %>% --> <!-- # mutate("#chrom" = chrom) --> <!-- temp = readr::read_tsv("filtered_id80_chr11_chr13_inDuplicatedArea_withGeneInfo.bed", col_names = F) --> <!-- colnames(temp)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- regions_withSD01MultiInShared = readr::read_tsv("../windowAnalysis/regions_withSD01MultiInShared.bed") --> <!-- new_regions_withSD01MultiInShared = readr::read_tsv("/Users/nick/projects/plasmodium/falciparum/PathWeaverAnalys_2020_12/chr11_chr13_inDuplicatedArea/PfSD01_chr11_chr13_inDuplicatedArea/final/hold.bed", col_names = F) --> <!-- colnames(new_regions_withSD01MultiInShared)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- firstPassSuccess = readr::read_tsv("chrom11_13DuplicatedRegion_successfulFirstPass.bed", col_names = F) --> <!-- colnames(firstPassSuccess)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- varWindowFromFirstPassSuccess = readr::read_tsv("finalTunned_duplicatedRegionChr11Chr13_combinedVarConservedRegions.bed", col_names = F) --> <!-- colnames(varWindowFromFirstPassSuccess)[1:length(colnames(outBed))] = colnames(outBed) --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen-inset-shaded --> <!-- overlayPlot = ggplot() + --> <!-- geom_vline(aes(xintercept = realPosition), --> <!-- data = variableSitesInPacbioGenomes, --> <!-- color = "red") + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = 0.5, ymax = 1), --> <!-- data = outBed) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = 1, ymax = 2), --> <!-- data = genomic_combined_filtered_3d7) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = 2, ymax = 3), fill = "maroon", --> <!-- data = inbetweenLargeTandems_inSharedRegion) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = 3, ymax = 4), fill = "darkblue", --> <!-- data = temp ) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = 3.75, ymax = 4), fill = "#00FCCF", --> <!-- data = firstPassSuccess ) + --> <!-- # geom_rect(aes(xmin = start, xmax = end, --> <!-- # ymin = 4, ymax = 5), fill = "darkblue", --> <!-- # data = temp2 ) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = 3, ymax = 3.5), fill = "limegreen", --> <!-- data = regions_withSD01MultiInShared ) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = 3, ymax = 3.25), fill = "forestgreen", --> <!-- data = new_regions_withSD01MultiInShared ) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = -1, ymax = -0.5), --> <!-- data = finalWindows_inSharedRegion) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = -0.5, ymax = 0), fill = "orange", --> <!-- data = finalSubWindows_inSharedRegion) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = 4, ymax = 5), fill = "orange", --> <!-- data = finalSubWindows_inSharedRegion) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = 0, ymax = 1, --> <!-- fill = description), --> <!-- data = allGenes_inSharedRegion) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = -0.75, ymax = -0.25), fill = "blue", --> <!-- data = combined_11_13_sharedRegions_stepped)+ --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = -0.75, ymax = -0.5), fill = "red", --> <!-- data = renamed_combined_sharedRegionsNotInTandems)+ --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = -1, ymax = -2), fill = "purple", --> <!-- data = inbetweenConservedRegions) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = -2.5, ymax = -2), fill = "lightblue", --> <!-- data = bedsOfSharedRegions)+ --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = -3, ymax = -2.5), fill = "magenta", --> <!-- data = sharedRegionsNotInTandems_size150_step25) + --> <!-- geom_rect(aes(xmin = start, xmax = end, --> <!-- ymin = 4, ymax = 4.25), fill = "#000000", --> <!-- data = varWindowFromFirstPassSuccess )+ --> <!-- scale_fill_manual(values = descriptionColors) + --> <!-- facet_wrap(~`#chrom`, scales = "free", ncol = 1) + --> <!-- sofonias_theme --> <!-- print(overlayPlot) --> <!-- ``` --> <!-- ```{r} --> <!-- #| column: screen-inset-shaded --> <!-- ggplotly(overlayPlot) --> <!-- ``` -->