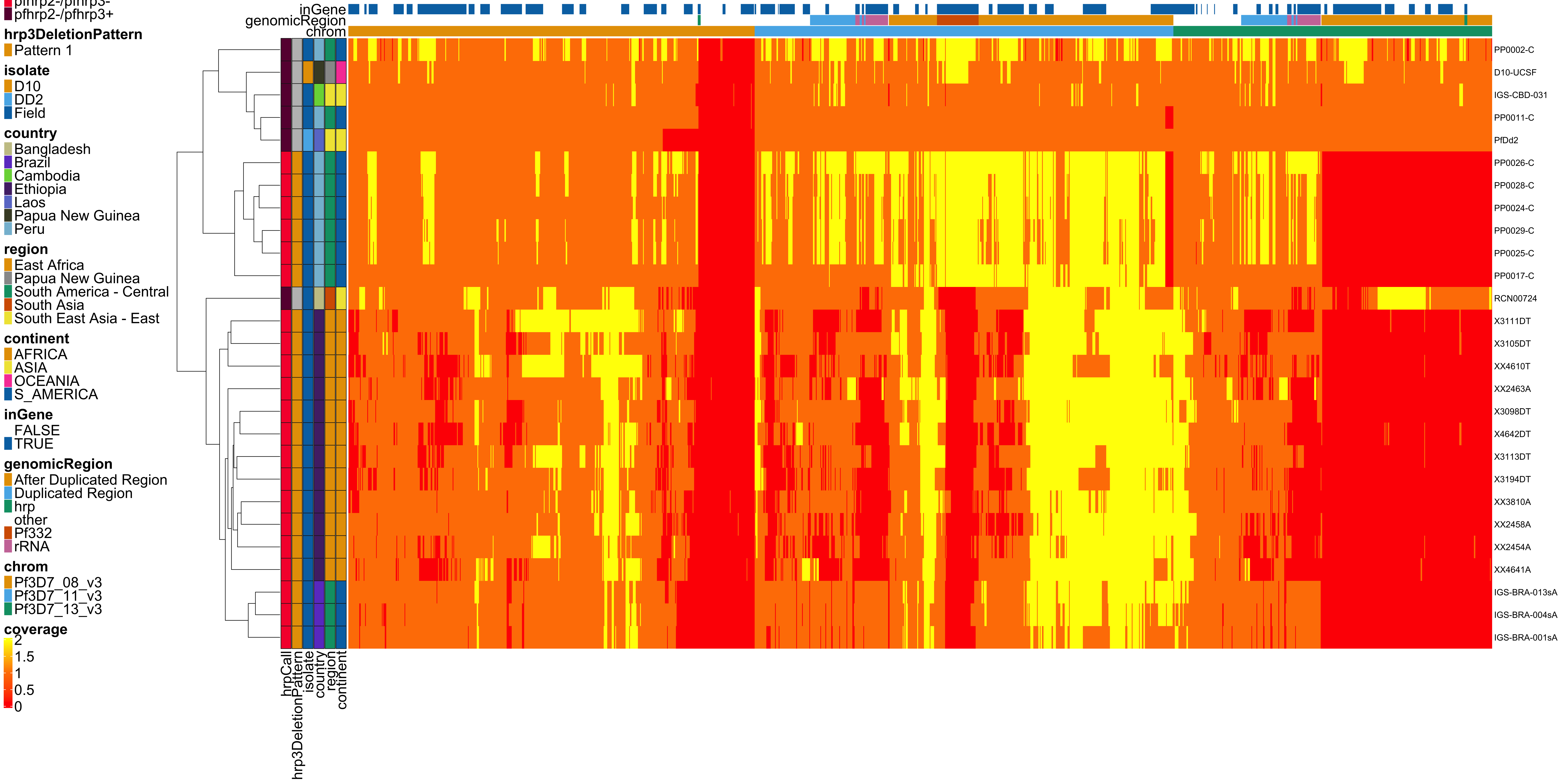
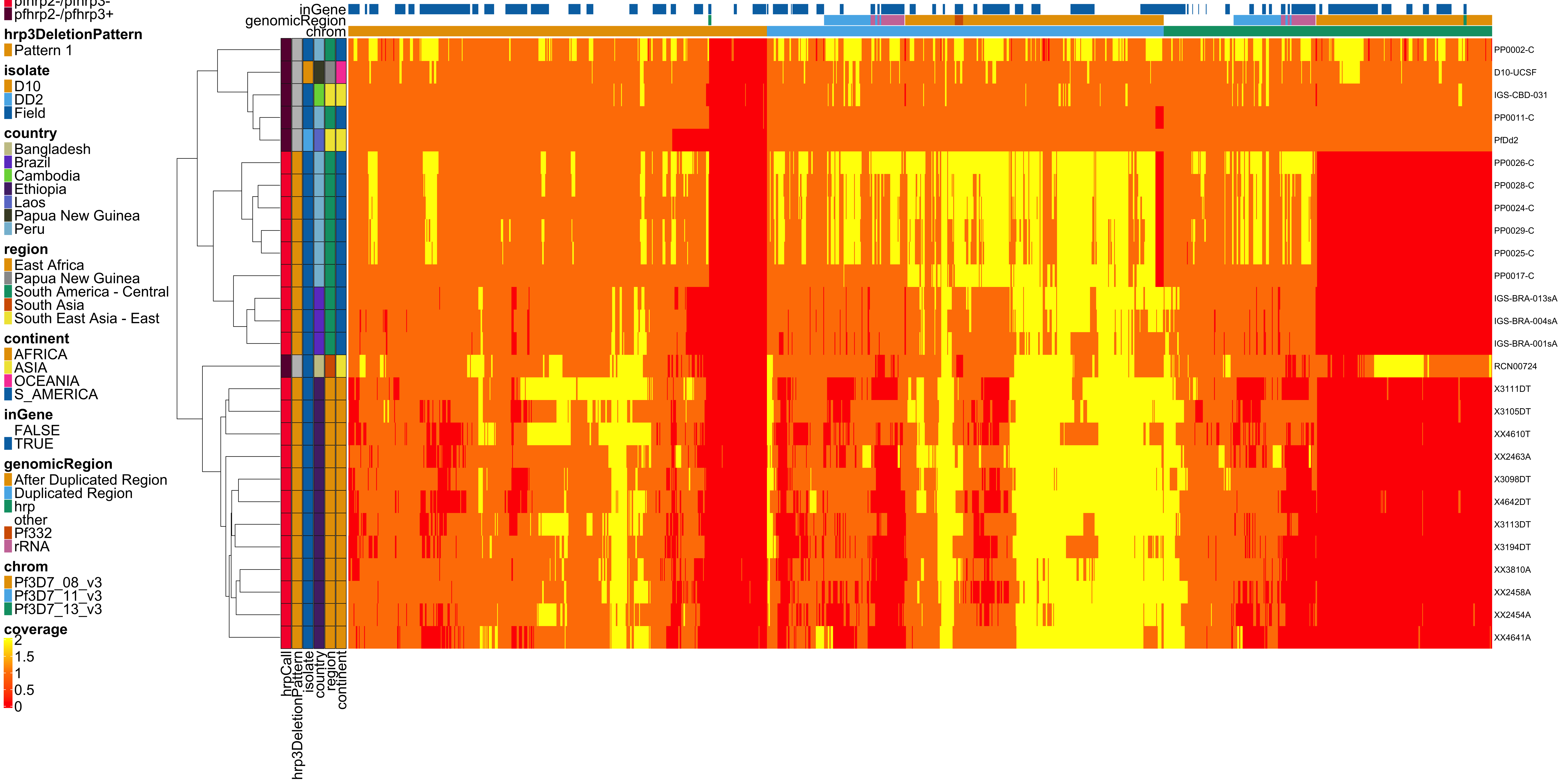
--- title: "Plotting coverage in sub windows" --- ```{r setup, echo=FALSE, message=FALSE} source("../common.R") ``` ```{r, echo=FALSE, eval=FALSE} metaByBioSample = readr::read_tsv("../meta/metadata/metaByBioSample.tab.txt") %>% mutate(country = gsub("South East Asia - East", "Cambodia", country)) metaByBioSample_Ethiopia = metaByBioSample %>% filter(country == "Ethiopia") allSel = readr::read_tsv("/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/pfepipanels/Pf_Epi_Panels/data/MAD4HATTER/data/pf/reports/slim_allSelectedClustersInfo.tab.txt.gz") allSel_xx = allSel %>% filter(grepl("^X",s_Sample) & s_Sample %in% metaByBioSample_Ethiopia$sample) allSel_xx_prep = HaplotypeRainbows::prepForRainbow(allSel_xx) HaplotypeRainbows::genRainbowHapPlotObj(allSel_xx_prep) allSel = readr::read_tsv("/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/pfepipanels/Pf_Epi_Panels/data/PfSMART/data/pf/reports/slim_allSelectedClustersInfo.tab.txt.gz") allSel_xx = allSel %>% filter(grepl("^X",s_Sample) & s_Sample %in% metaByBioSample_Ethiopia$sample) allSel_xx_prep = HaplotypeRainbows::prepForRainbow(allSel_xx, minPopSize = 1) HaplotypeRainbows::genRainbowHapPlotObj(allSel_xx_prep) allSel = readr::read_tsv("/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/pfepipanels/Pf_Epi_Panels/data/heome1/data/pf/reports/slim_allSelectedClustersInfo.tab.txt.gz") allSel_withDeletions = allSel %>% filter(s_Sample %in% previousDeletionCalls$BiologicalSample) allSel_withDeletions_prep = HaplotypeRainbows::prepForRainbow(allSel_withDeletions, minPopSize = 2) pdf("heome1_rainbow.pdf", width = 20, height = 25, useDingbats = F) HaplotypeRainbows::genRainbowHapPlotObj(allSel_withDeletions_prep) dev.off() allSel_withDeletions_prep_outForMoire = allSel_withDeletions_prep %>% select(s_Sample, p_name, h_popUID) %>% rename(sample_id = s_Sample, locus = p_name, allele = h_popUID) write_tsv(allSel_withDeletions_prep_outForMoire, "~/Downloads/allSel_withDeletions_prep_outForMoire.tsv") ``` ```{r} = readr:: read_tsv ("../meta/metadata/meta.tab.txt" ) %>% mutate (country = gsub ("South East Asia - East" , "Cambodia" , country))= readr:: read_tsv ("../meta/metadata/metaByBioSample.tab.txt" ) %>% mutate (country = gsub ("South East Asia - East" , "Cambodia" , country))# coiCalls = readr::read_tsv("MAD4HATTER_COI_calls.tab.txt") # #coiCalls = readr::read_tsv("PfSMART_COI_calls.tab.txt") # coiCalls = readr::read_tsv("heome1_COI_calls.tab.txt") # # # coiCalls_poly = coiCalls %>% # filter(COI > 1) = readr:: read_tsv ("allMeta_HRP2_HRP3_deletionCalls.tab.txt" ) %>% #filter(country %!in% c("Bangladesh", "Mauritania", "Myanmar", "The Gambia")) %>% filter (! ((grepl ("SPT" , sample) & possiblyChr11Deleted))) %>% #filter(BiologicalSample %!in% coiCalls_poly$sample) %>% mutate (country = gsub ("South East Asia - East" , "Cambodia" , country)) %>% mutate (hrpCall = case_when (& possiblyHRP3Deleted ~ "pfhrp2-/pfhrp3-" , & ! possiblyHRP3Deleted ~ "pfhrp2-/pfhrp3+" , ! possiblyHRP2Deleted & possiblyHRP3Deleted ~ "pfhrp2+/pfhrp3-" , ~ "pfhrp2+/pfhrp3+" # %>% # left_join(coiCalls %>% # rename(BiologicalSample = sample)) = previousDeletionCalls %>% filter (possiblyHRP2Deleted)= previousDeletionCalls %>% filter (possiblyHRP3Deleted)= previousDeletionCalls %>% filter (possiblyChr11Deleted)= readr:: read_tsv ("../meta/metadata/masterTable.tab.txt" ) %>% filter (! is.na (SRARuns))= readr:: read_tsv ("../meta/allCov_summaryStats.tab.txt.gz" )#cov = readr::read_tsv("../../../../allSRAData/reProcess_2021_11_19/coverage/data/allCov_summaryStats.tab.txt.gz") = meta%>% left_join (previousDeletionCalls)%>% mutate (isolate = ifelse (IsFieldSample, "Field" , BiologicalSample))= "data/finalHRPII_HRPIII_windows_withTunedSubWindows/popClustering/reports/allBasicInfo.tab.txt.gz" = "finalHRPII_HRPIII_windows_withTunedSubWindows_allBasicInfo.Rdata" if (! file.exists (outputFnp) | file.info (inputFnp)$ ctime > file.info (outputFnp)$ ctime){= readr:: read_tsv (inputFnp)%>% mutate (genomicID = paste0 (` #chrom ` , "-" , start, "-" , end))save (allBasicInfo, file = outputFnp)else {load (outputFnp)= readr:: read_tsv ("../rRNA_segmental_duplications/sharedBetween11_and_13/investigatingChrom11Chrom13/Pf3D7_13_v3-2792021-2807295-for--Pf3D7_11_v3-1918028-1933288-for.bed" , col_names = F)= allBasicInfo %>% filter (sample %fin% previousDeletionCalls$ sample) %>% # filter(name %!in% Chrom13RegionsToRemove) %>% #mutate(inGene = !is.na(extraField0)) %>% mutate (inGene = extraField0 != "[extraField0=NA]" ) %>% left_join (cov) %>% mutate (medianCov = median (perBaseCoverage), meanCov = mean (perBaseCoverage)) %>% mutate (perBaseCoverageNorm = ifelse (inGene, perBaseCoverage/ medianPerBaseCov_inGenes, perBaseCoverage/ medianPerBaseCov_notInGenes)) %>% mutate (perBaseCoverageNormRounded = ifelse (perBaseCoverageNorm > 0.10 & perBaseCoverageNorm <= 1 , 1 , perBaseCoverageNorm)) %>% mutate (perBaseCoverageNormRounded = round (perBaseCoverageNormRounded)) # mutate(perBaseCoverageNormRounded = ifelse(perBaseCoverageNorm >0.10 & perBaseCoverageNorm < 0.5, 0.5, perBaseCoverageNorm)) %>% # mutate(perBaseCoverageNormRounded = round(perBaseCoverageNormRounded/0.5)*0.5) = allBasicInfo_filt %>% filter (.$ sample[1 ] == sample) %>% select (1 : 6 , extraField0) %>% unique () %>% mutate (genomicID = paste0 (` #chrom ` , "-" , start, "-" , end)) = regions %>% mutate (id = paste0 (` #chrom ` , "-" , start, "-" , end)) %>% arrange (id) %>% #mutate(inGene = !is.na(extraField0)) %>% mutate (inGene = extraField0 != "[extraField0=NA]" ) %>% mutate (geneType = ifelse (grepl ("histidine-rich protein II" , regions$ extraField0),"hrp" ,"other" %>% mutate (geneType = ifelse (grepl ("ribosomal RNA" , regions$ extraField0), "rRNA" , geneType)) %>% mutate (geneType = ifelse (grepl ("332" , regions$ extraField0), "Pf332" , geneType)) %>% mutate (homologousRegion = ifelse ((` #chrom ` == "Pf3D7_11_v3" & >= 1918028 & <= 1933288 ) | ` #chrom ` == "Pf3D7_13_v3" & >= 2792021 & <= 2807295 ,"shared" ,"other" %>% mutate (afterHomologousRegion = (` #chrom ` == "Pf3D7_11_v3" & >= 1933288 ) | ` #chrom ` == "Pf3D7_13_v3" & >= 2807295 )) %>% mutate (genomicRegion = case_when ("rRNA" == geneType ~ "rRNA" ,"hrp" == geneType ~ "hrp" ,"Pf332" == geneType ~ "Pf332" ,~ "After Duplicated Region" , "shared" == homologousRegion ~ "Duplicated Region" , ~ "other" %>% mutate (chrom = ` #chrom ` )## further filtering = c ("Pf3D7_08_v3-1375207-1375341__var-0" ,"Pf3D7_08_v3-1375210-1375410__var-0" ,"Pf3D7_08_v3-1375185-1375485__var-1" ,"Pf3D7_08_v3-1375557-1375750__subseq-0" ,"Pf3D7_08_v3-1378006-1378662__var-0" ,"Pf3D7_08_v3-1378006-1378662__var-1" ,"Pf3D7_08_v3-1378006-1378662__var-2" ,"Pf3D7_08_v3-1378006-1378662__var-3" ,"Pf3D7_08_v3-1378006-1378662__var-4" ,"Pf3D7_08_v3-1379154-1379915__var-0" ,"Pf3D7_08_v3-1379154-1379915__var-1" ,"Pf3D7_08_v3-1379154-1379915__var-2" ,"Pf3D7_08_v3-1379154-1379915__var-3" ,"Pf3D7_08_v3-1379154-1379915__var-4" ,"Pf3D7_08_v3-1379154-1379915__var-5" ,"Pf3D7_08_v3-1379154-1379915__var-6" ,"Pf3D7_08_v3-1379154-1379915__var-7" ,"Pf3D7_08_v3-1379154-1379915__var-8" ,"Pf3D7_08_v3-1380194-1380328__var-0" ,"Pf3D7_08_v3-1382255-1382505__var-0" ,"Pf3D7_08_v3-1382255-1382505__var-1" ,"Pf3D7_08_v3-1382255-1382505__var-2" ,"Pf3D7_08_v3-1382680-1383155__var-0" ,"Pf3D7_08_v3-1382680-1383155__var-1" ,"Pf3D7_08_v3-1382680-1383155__var-2" ,"Pf3D7_08_v3-1382680-1383155__var-3" ,"Pf3D7_08_v3-1382680-1383155__var-4" ,"Pf3D7_08_v3-1382680-1383155__var-5" ,"Pf3D7_08_v3-1382680-1383155__var-6" ,"Pf3D7_08_v3-1382680-1383155__var-7" ,"Pf3D7_08_v3-1384030-1384251__var-0" ,"Pf3D7_08_v3-1384030-1384251__var-1" ,"Pf3D7_08_v3-1384030-1384251__var-2" ,"Pf3D7_08_v3-1384316-1384663__var-0" ,"Pf3D7_08_v3-1384316-1384663__var-1" ,"Pf3D7_08_v3-1384316-1384663__var-2" ,"Pf3D7_08_v3-1384316-1384663__var-3" ,"Pf3D7_08_v3-1384316-1384663__var-4" ,"Pf3D7_08_v3-1384837-1385370__var-0" ,"Pf3D7_08_v3-1384837-1385370__var-1" ,"Pf3D7_08_v3-1384837-1385370__var-2" ,"Pf3D7_08_v3-1384837-1385370__var-3" ,"Pf3D7_08_v3-1384837-1385370__var-4" ,"Pf3D7_08_v3-1384837-1385370__var-5" ,"Pf3D7_08_v3-1384837-1385370__var-6" ,"Pf3D7_08_v3-1385625-1385741__var-0" ,"Pf3D7_08_v3-1385951-1386323__var-0" ,"Pf3D7_08_v3-1385951-1386323__var-1" ,"Pf3D7_08_v3-1385951-1386323__var-2" ,"Pf3D7_08_v3-1385951-1386323__var-3" ,"Pf3D7_08_v3-1385951-1386323__var-4" ,"Pf3D7_08_v3-1385951-1386323__var-5" ,"Pf3D7_08_v3-1386518-1386680__var-0" ,"Pf3D7_08_v3-1386518-1386680__var-1" ,"Pf3D7_08_v3-1386518-1386680__var-2" ,"Pf3D7_08_v3-1386518-1386680__var-3" ,"Pf3D7_08_v3-1386739-1387414__var-00" ,"Pf3D7_08_v3-1386739-1387414__var-01" ,"Pf3D7_08_v3-1386739-1387414__var-02" ,"Pf3D7_08_v3-1386739-1387414__var-03" ,"Pf3D7_08_v3-1386739-1387414__var-04" ,"Pf3D7_08_v3-1386739-1387414__var-05" ,"Pf3D7_08_v3-1386739-1387414__var-06" ,"Pf3D7_08_v3-1386739-1387414__var-07" ,"Pf3D7_08_v3-1386739-1387414__var-08" ,"Pf3D7_08_v3-1386739-1387414__var-09" ,"Pf3D7_08_v3-1386739-1387414__var-10" ,"Pf3D7_08_v3-1386739-1387414__var-11" ,"Pf3D7_08_v3-1386739-1387414__var-12" ,"Pf3D7_08_v3-1386739-1387414__var-13" ,"Pf3D7_08_v3-1387782-1387982__var-0" ,"Pf3D7_08_v3-1387782-1387982__var-1" ,"Pf3D7_08_v3-1387782-1387982__var-2" ,"Pf3D7_08_v3-1387782-1387982__var-3" )= c ("Pf3D7_11_v3-1991347-1992851__var-00" ,"Pf3D7_11_v3-1991347-1992851__var-01" ,"Pf3D7_11_v3-1991347-1992851__var-02" ,"Pf3D7_11_v3-1991347-1992851__var-03" ,"Pf3D7_11_v3-1991347-1992851__var-04" ,"Pf3D7_11_v3-1991347-1992851__var-05" ,"Pf3D7_11_v3-1991347-1992851__var-06" ,"Pf3D7_11_v3-1991347-1992851__var-07" ,"Pf3D7_11_v3-1991347-1992851__var-08" ,"Pf3D7_11_v3-1991347-1992851__var-09" ,"Pf3D7_11_v3-1991347-1992851__var-10" ,"Pf3D7_11_v3-1991347-1992851__var-11" ,"Pf3D7_11_v3-1991347-1992851__var-12" ,"Pf3D7_11_v3-1991347-1992851__var-13" ,"Pf3D7_11_v3-1991347-1992851__var-14" ,"Pf3D7_11_v3-1991347-1992851__var-15" ,"Pf3D7_11_v3-1991347-1992851__var-16" ,"Pf3D7_11_v3-1991347-1992851__var-17" ,"Pf3D7_11_v3-1991347-1992851__var-18" ,"Pf3D7_11_v3-1992907-1993669__var-00" ,"Pf3D7_11_v3-1992907-1993669__var-01" ,"Pf3D7_11_v3-1992907-1993669__var-02" ,"Pf3D7_11_v3-1992907-1993669__var-03" ,"Pf3D7_11_v3-1992907-1993669__var-04" ,"Pf3D7_11_v3-1992907-1993669__var-05" ,"Pf3D7_11_v3-1992907-1993669__var-06" ,"Pf3D7_11_v3-1992907-1993669__var-07" ,"Pf3D7_11_v3-1992907-1993669__var-08" ,"Pf3D7_11_v3-1992907-1993669__var-09" ,"Pf3D7_11_v3-1993833-1994061__var-0" ,"Pf3D7_11_v3-1993833-1994061__var-1" ,"Pf3D7_11_v3-1993833-1994061__var-2" ,"Pf3D7_11_v3-1993833-1994061__var-3" ,"Pf3D7_11_v3-1994139-1994363__var-0" ,"Pf3D7_11_v3-1994139-1994363__var-1" ,"Pf3D7_11_v3-1994139-1994363__var-2" ,"Pf3D7_11_v3-1994139-1994363__var-3" ,"Pf3D7_11_v3-1995234-1995394__var-0" ,"Pf3D7_11_v3-1995234-1995394__var-1" ,"Pf3D7_11_v3-1995459-1995614__var-0" ,"Pf3D7_11_v3-1995459-1995614__var-1" ,"Pf3D7_11_v3-1995459-1995614__var-2" ,"Pf3D7_11_v3-1995678-1996112__var-00" ,"Pf3D7_11_v3-1995678-1996112__var-01" ,"Pf3D7_11_v3-1995678-1996112__var-02" ,"Pf3D7_11_v3-1995678-1996112__var-03" ,"Pf3D7_11_v3-1995678-1996112__var-04" ,"Pf3D7_11_v3-1995678-1996112__var-05" ,"Pf3D7_11_v3-1995678-1996112__var-06" ,"Pf3D7_11_v3-1995678-1996112__var-07" ,"Pf3D7_11_v3-1995678-1996112__var-08" ,"Pf3D7_11_v3-1995678-1996112__var-09" ,"Pf3D7_11_v3-1996349-1996650__var-0" ,"Pf3D7_11_v3-1996349-1996650__var-1" ,"Pf3D7_11_v3-1996745-1997019__var-0" ,"Pf3D7_11_v3-1996745-1997019__var-1" ,"Pf3D7_11_v3-1996745-1997019__var-2" ,"Pf3D7_11_v3-1996745-1997019__var-3" ,"Pf3D7_11_v3-1996745-1997019__var-4" ,"Pf3D7_11_v3-1997095-1997221__var-0" ,"Pf3D7_11_v3-1997095-1997221__var-1" ,"Pf3D7_11_v3-1997095-1997221__var-2" ,"Pf3D7_11_v3-1997095-1997221__var-3" ,"Pf3D7_11_v3-1997276-1997425__var-0" ,"Pf3D7_11_v3-1997276-1997425__var-1" ,"Pf3D7_11_v3-1997276-1997425__var-2" ,"Pf3D7_11_v3-1997276-1997425__var-3" ,"Pf3D7_11_v3-1997674-1997884__var-0" ,"Pf3D7_11_v3-1997674-1997884__var-1" ,"Pf3D7_11_v3-1997944-1998371__var-0" ,"Pf3D7_11_v3-1997944-1998371__var-1" ,"Pf3D7_11_v3-1997944-1998371__var-2" ,"Pf3D7_11_v3-1997944-1998371__var-3" ,"Pf3D7_11_v3-1997944-1998371__var-4" ,"Pf3D7_11_v3-1997944-1998371__var-5" ,"Pf3D7_11_v3-1997944-1998371__var-6" ,"Pf3D7_11_v3-1997944-1998371__var-7" ,"Pf3D7_11_v3-1998638-1998759__var-0" ,"Pf3D7_11_v3-1998833-1999151__var-0" ,"Pf3D7_11_v3-1998833-1999151__var-1" ,"Pf3D7_11_v3-1998833-1999151__var-2" ,"Pf3D7_11_v3-1998833-1999151__var-3" ,"Pf3D7_11_v3-1999245-1999390__var-0" ,"Pf3D7_11_v3-1999600-1999713__var-0" ,"Pf3D7_11_v3-1999600-1999713__var-1" ,"Pf3D7_11_v3-2000687-2000889__var-0" ,"Pf3D7_11_v3-2000687-2000889__var-1" ,"Pf3D7_11_v3-2000687-2000889__var-2" ,"Pf3D7_11_v3-2000687-2000889__var-3" ,"Pf3D7_11_v3-2000687-2000889__var-4" ,"Pf3D7_11_v3-2000687-2000889__var-5" ,"Pf3D7_11_v3-2000941-2001239__var-0" ,"Pf3D7_11_v3-2000941-2001239__var-1" ,"Pf3D7_11_v3-2000941-2001239__var-2" ,"Pf3D7_11_v3-2000941-2001239__var-3" ,"Pf3D7_11_v3-2000941-2001239__var-4" ,"Pf3D7_11_v3-2000941-2001239__var-5" ,"Pf3D7_11_v3-2001300-2003328__var-00" ,"Pf3D7_11_v3-2001300-2003328__var-01" ,"Pf3D7_11_v3-2001300-2003328__var-02" ,"Pf3D7_11_v3-2001300-2003328__var-03" ,"Pf3D7_11_v3-2001300-2003328__var-04" ,"Pf3D7_11_v3-2001300-2003328__var-05" ,"Pf3D7_11_v3-2001300-2003328__var-06" ,"Pf3D7_11_v3-2001300-2003328__var-07" ,"Pf3D7_11_v3-2001300-2003328__var-08" ,"Pf3D7_11_v3-2001300-2003328__var-09" ,"Pf3D7_11_v3-2001300-2003328__var-10" ,"Pf3D7_11_v3-2001300-2003328__var-11" ,"Pf3D7_11_v3-2001300-2003328__var-12" ,"Pf3D7_11_v3-2001300-2003328__var-13" ,"Pf3D7_11_v3-2001300-2003328__var-14" ,"Pf3D7_11_v3-2001300-2003328__var-15" ,"Pf3D7_11_v3-2001300-2003328__var-16" ,"Pf3D7_11_v3-2001300-2003328__var-17" ,"Pf3D7_11_v3-2001300-2003328__var-18" ,"Pf3D7_11_v3-2001300-2003328__var-19" ,"Pf3D7_11_v3-2001300-2003328__var-20" ,"Pf3D7_11_v3-2001300-2003328__var-21" ,"Pf3D7_11_v3-2001300-2003328__var-22" ,"Pf3D7_11_v3-2001300-2003328__var-23" ,"Pf3D7_11_v3-2001300-2003328__var-24" ,"Pf3D7_11_v3-2001300-2003328__var-25" ,"Pf3D7_11_v3-2001300-2003328__var-26" )= c ("Pf3D7_13_v3-2841776-2841926__subseq-0" ,"Pf3D7_13_v3-2842001-2842301__var-0" ,"Pf3D7_13_v3-2842076-2842151__var-0" ,"Pf3D7_13_v3-2842076-2842176__var-0" ,"Pf3D7_13_v3-2842101-2842251__var-0" ,"Pf3D7_13_v3-2842001-2842301__var-1" ,"Pf3D7_13_v3-2842151-2842226__var-0" ,"Pf3D7_13_v3-2842226-2842301__var-0" ,"Pf3D7_13_v3-2842001-2842301__var-2" ,"Pf3D7_13_v3-2842251-2842351__var-0" ,"Pf3D7_13_v3-2842301-2842376__var-0" ,"Pf3D7_13_v3-2842251-2842401__var-0" ,"Pf3D7_13_v3-2842251-2842451__var-0" ,"Pf3D7_13_v3-2842426-2842501__var-0" ,"Pf3D7_13_v3-2842476-2842688__var-0" ,"Pf3D7_13_v3-2842476-2842688__var-1" ,"Pf3D7_13_v3-2842476-2842688__var-2" ,"Pf3D7_13_v3-2842515-2842804__var-2" ,"Pf3D7_13_v3-2843108-2843446__var-0" ,"Pf3D7_13_v3-2843108-2843446__var-1" ,"Pf3D7_13_v3-2843108-2843446__var-2" ,"Pf3D7_13_v3-2843108-2843446__var-3" ,"Pf3D7_13_v3-2843108-2843446__var-4" ,"Pf3D7_13_v3-2843108-2843446__var-5" ,"Pf3D7_13_v3-2843641-2843863__var-0" ,"Pf3D7_13_v3-2843641-2843863__var-1" ,"Pf3D7_13_v3-2843930-2844101__var-0" ,"Pf3D7_13_v3-2843930-2844101__var-1" ,"Pf3D7_13_v3-2844247-2844785__var-0" ,"Pf3D7_13_v3-2844247-2844785__var-1" ,"Pf3D7_13_v3-2844247-2844785__var-2" ,"Pf3D7_13_v3-2844247-2844785__var-3" ,"Pf3D7_13_v3-2844247-2844785__var-4" ,"Pf3D7_13_v3-2844247-2844785__var-5" )= readr:: read_tsv ("/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/hrps/windowSelectionSurroundingHRPs/Windows_Surrounding_HRP2_3_deletions/windowAnalysis/allMeta_HRP2_HRP3_deletionCalls.tab.txt" #### filter ends chr08 -begin = allBasicInfo_filt %>% filter (` #chrom ` == "Pf3D7_08_v3" ) %>% filter (%in% previousDeletionCalls_hrp2_del$ sample%>% group_by (sample, name) %>% mutate (marker = ifelse (perBaseCoverageNormRounded == 0 , 1 , 0 )) %>% filter (name %in% endRegions_08) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker)) %>% mutate (noCovPerc = noCov/ length (endRegions_08))= allBasicInfo_filt_endsNoCovSel_08 %>% #filter(noCov == length(endRegions_08)) filter (noCovPerc >= 0.95 )= allBasicInfo_filt %>% filter (` #chrom ` == "Pf3D7_08_v3" ) %>% filter (%in% previousDeletionCalls_hrp2_del$ sample%>% group_by (sample, name) %>% mutate (marker = ifelse (readTotal > 10 & success, 0 , 1 )) %>% filter (name %in% endRegions_08) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker))%>% mutate (noCovPerc = noCov/ length (endRegions_08))= allBasicInfo_filt_endsNoSuccessSel_08 %>% #filter(noCov == length(endRegions_08)) filter (noCovPerc >= 0.95 )= intersect (sort ($ samplesort ($ sample#### filter ends chr08 -end #### filter ends chr13 -begin = allBasicInfo_filt %>% filter (` #chrom ` == "Pf3D7_13_v3" ) %>% filter (%in% previousDeletionCalls_hrp3_del$ sample%>% group_by (sample, name) %>% mutate (marker = ifelse (perBaseCoverageNormRounded == 0 , 1 , 0 )) %>% filter (name %in% endRegions_13) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker)) %>% mutate (noCovPerc = noCov/ length (endRegions_13))= allBasicInfo_filt_endsNoCovSel_13 %>% #filter(noCov == length(endRegions_13)) filter (noCovPerc >= 0.95 )= allBasicInfo_filt %>% filter (` #chrom ` == "Pf3D7_13_v3" ) %>% filter (%in% previousDeletionCalls_hrp3_del$ sample%>% group_by (sample, name) %>% mutate (marker = ifelse (readTotal > 10 & success, 0 , 1 )) %>% filter (name %in% endRegions_13) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker))%>% mutate (noCovPerc = noCov/ length (endRegions_13))= allBasicInfo_filt_endsNoSuccessSel_13 %>% #filter(noCov == length(endRegions_13)) filter (noCovPerc >= 0.95 )= intersect (sort ($ samplesort ($ sample#### filter ends chr13 -end #### filter ends chr11 -begin = allBasicInfo_filt %>% filter (` #chrom ` == "Pf3D7_11_v3" ) %>% filter (%in% previousDeletionCalls_chr11_del$ sample%>% group_by (sample, name) %>% mutate (marker = ifelse (perBaseCoverageNormRounded == 0 , 1 , 0 )) %>% filter (name %in% endRegions_11) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker)) %>% mutate (noCovPerc = noCov/ length (endRegions_11))= allBasicInfo_filt_endsNoCovSel_11 %>% #filter(noCov == length(endRegions_11)) filter (noCovPerc >= 0.95 )= allBasicInfo_filt %>% filter (` #chrom ` == "Pf3D7_11_v3" ) %>% filter (%in% previousDeletionCalls_chr11_del$ sample%>% group_by (sample, name) %>% mutate (marker = ifelse (readTotal > 10 & success, 0 , 1 )) %>% filter (name %in% endRegions_11) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker))%>% mutate (noCovPerc = noCov/ length (endRegions_11))= allBasicInfo_filt_endsNoSuccessSel_11 %>% #filter(noCov == length(endRegions_11)) filter (noCovPerc >= 0.95 )= intersect (sort ($ samplesort ($ sample#### filter ends chr11 -end = c (%>% unique ()= allBasicInfo_filt %>% select (sample) %>% unique () %>% filter (sample %!in% samplesToKeep)= readr:: read_tsv ("initial_allMeta_HRP2_HRP3_deletionCalls.tab.txt" ) %>% filter (sample %in% samplesToKeep) %>% #filter(country %!in% c("Bangladesh", "Mauritania", "Myanmar", "The Gambia")) %>% filter (! ((grepl ("SPT" , sample) & possiblyChr11Deleted))) %>% #filter(BiologicalSample %!in% coiCalls_poly$sample) %>% mutate (country = gsub ("South East Asia - East" , "Cambodia" , country)) %>% mutate (hrpCall = case_when (& possiblyHRP3Deleted ~ "pfhrp2-/pfhrp3-" , & ! possiblyHRP3Deleted ~ "pfhrp2-/pfhrp3+" , ! possiblyHRP2Deleted & possiblyHRP3Deleted ~ "pfhrp2+/pfhrp3-" , ~ "pfhrp2+/pfhrp3+" write_tsv (previousDeletionCalls, "allMeta_HRP2_HRP3_deletionCalls.tab.txt" )= previousDeletionCalls %>% filter (possiblyHRP2Deleted)= previousDeletionCalls %>% filter (possiblyHRP3Deleted)= previousDeletionCalls %>% filter (possiblyChr11Deleted)= allBasicInfo_filt %>% filter (sample %in% samplesToKeep)= allBasicInfo_filt %>% group_by () %>% select (sample, genomicID, perBaseCoverageNormRounded) %>% spread (genomicID, perBaseCoverageNormRounded)= as.matrix (allBasicInfo_filt_sp[,2 : ncol (allBasicInfo_filt_sp)])rownames (allBasicInfo_filt_sp_mat) = allBasicInfo_filt_sp$ sample> 2 ] = 2 = regions[match (colnames (allBasicInfo_filt_sp_mat), regions$ genomicID),]= meta[match (allBasicInfo_filt_sp$ sample, meta$ sample), ]= metaSelected %>% filter (possiblyHRP2Deleted)= metaSelected %>% filter (possiblyHRP3Deleted)= metaSelected %>% filter (possiblyHRP2Deleted, possiblyHRP3Deleted)= regions %>% select (name, genomicID)``` ## Writing out deletion calls ```{r} = allBasicInfo_filt %>% left_join (regions)= allBasicInfo_filt_counting %>% group_by (sample, ` #chrom ` , afterHomologousRegion) %>% mutate (marker = perBaseCoverageNormRounded > 0 ) %>% summarise (markerSum = sum (marker), n = n ()) %>% mutate (markerFrac = markerSum/ n) %>% filter (afterHomologousRegion)= allBasicInfo_filt_counting_sum %>% group_by () %>% select (sample, ` #chrom ` , markerFrac) %>% spread (` #chrom ` , markerFrac) %>% mutate ()= allBasicInfo_filt_counting_sum_sp %>% filter (sample %in% metaSelected_hrp3_deleted$ sample) %>% mutate (HRP3_deletionPattern = ifelse (Pf3D7_13_v3 < 0.10 , "Pattern 1" , "Pattern 2" ))= previousDeletionCalls %>% left_join (allBasicInfo_filt_counting_sum_sp_hrp3Deleted %>% group_by () %>% select (HRP3_deletionPattern, sample))write_tsv (previousDeletionCalls %>% #filter(hrpCall != "pfhrp2+/pfhrp3+") %>% left_join (masterTable %>% select (sample, SRARuns)), file = "allMetaDeletionCalls.tab.txt" )write_tsv (previousDeletionCalls %>% #filter(hrpCall != "pfhrp2+/pfhrp3+") %>% left_join (masterTable %>% select (sample, SRARuns)) %>% filter ("Pattern 2" == HRP3_deletionPattern), file = "allMetaDeletionCalls_Hrp3pattern2.tab.txt" )``` ```{r} # counts per country = previousDeletionCalls %>% filter (hrpCall != "pfhrp2+/pfhrp3+" ) %>% filter (IsFieldSample) %>% group_by (country, region, secondaryRegion, hrpCall) %>% count ()= previousDeletionCalls %>% filter (! is.na (HRP3_deletionPattern)) %>% filter (IsFieldSample) %>% group_by (country, region, secondaryRegion, HRP3_deletionPattern) %>% count ()write_tsv (previousDeletionCalls_countryHRPCallSum, "HRP_deletion_calls_by_country.tab.txt" )write_tsv (previousDeletionCalls_countryHRP3CallSum, "HRP3_deletionPattern_calls_by_country.tab.txt" )# counts per region = previousDeletionCalls %>% filter (hrpCall != "pfhrp2+/pfhrp3+" ) %>% filter (IsFieldSample) %>% group_by (region, secondaryRegion, hrpCall) %>% count ()= previousDeletionCalls %>% filter (! is.na (HRP3_deletionPattern)) %>% filter (IsFieldSample) %>% group_by (region, secondaryRegion, HRP3_deletionPattern) %>% count ()write_tsv (previousDeletionCalls_regionHRPCallSum, "HRP_deletion_calls_by_region.tab.txt" )write_tsv (previousDeletionCalls_regionHRP3CallSum, "HRP3_deletionPattern_calls_by_region.tab.txt" )# counter per continent = previousDeletionCalls %>% filter (hrpCall != "pfhrp2+/pfhrp3+" ) %>% filter (IsFieldSample) %>% group_by (secondaryRegion, hrpCall) %>% count ()= previousDeletionCalls %>% filter (! is.na (HRP3_deletionPattern)) %>% filter (IsFieldSample) %>% group_by (secondaryRegion, HRP3_deletionPattern) %>% count ()write_tsv (previousDeletionCalls_continentHRPCallSum, "HRP_deletion_calls_by_continent.tab.txt" )write_tsv (previousDeletionCalls_continentHRP3CallSum, "HRP3_deletionPattern_calls_by_continent.tab.txt" )# total = previousDeletionCalls %>% filter (hrpCall != "pfhrp2+/pfhrp3+" ) %>% filter (IsFieldSample) %>% group_by (hrpCall) %>% count ()= previousDeletionCalls %>% filter (! is.na (HRP3_deletionPattern)) %>% filter (IsFieldSample) %>% group_by (HRP3_deletionPattern) %>% count () %>% ungroup () %>% mutate (total = sum (n))write_tsv (previousDeletionCalls_HRPCallSum, "HRP_deletion_calls_total.tab.txt" )write_tsv (previousDeletionCalls_HRP3CallSum, "HRP3_deletionPattern_calls_total.tab.txt" )``` <!-- ### Counts by continent --> <!-- #### All --> <!-- ```{r} --> <!-- previousDeletionCalls_hrp2_del_continentSum = previousDeletionCalls %>% --> <!-- filter(possiblyHRP2Deleted) %>% --> <!-- group_by(secondaryRegion) %>% --> <!-- count(name = "hrp2_deletion_count") %>% --> <!-- ungroup() %>% --> <!-- mutate(total_hrp2_deletion_count = sum(hrp2_deletion_count)) --> <!-- previousDeletionCalls_hrp3_del_continentSum = previousDeletionCalls %>% --> <!-- filter(possiblyHRP3Deleted) %>% --> <!-- group_by(secondaryRegion) %>% --> <!-- count(name = "hrp3_deletion_count") %>% --> <!-- ungroup() %>% --> <!-- mutate(total_hrp3_deletion_count = sum(hrp3_deletion_count)) --> <!-- previousDeletionCalls_hrp2_and_hrp3_del_continentSum = previousDeletionCalls %>% --> <!-- filter(possiblyHRP2Deleted & possiblyHRP3Deleted) %>% --> <!-- group_by(secondaryRegion) %>% --> <!-- count(name = "hrp2_and_hrp3_deletion_count") %>% --> <!-- ungroup() %>% --> <!-- mutate(total_hrp2_and_hrp3_deletion_count = sum(hrp2_and_hrp3_deletion_count)) --> <!-- previousDeletionCalls_hrp2_hrp3_del_continentSum = previousDeletionCalls_hrp2_del_continentSum %>% --> <!-- full_join(previousDeletionCalls_hrp3_del_continentSum) %>% --> <!-- full_join(previousDeletionCalls_hrp2_and_hrp3_del_continentSum) %>% --> <!-- ungroup() %>% --> <!-- select(-starts_with("total")) %>% --> <!-- mutate_if(is.numeric,dplyr::coalesce,0) %>% --> <!-- mutate(total_hrp2_and_hrp3_deletion_count = sum(hrp2_and_hrp3_deletion_count)) %>% --> <!-- mutate(total_hrp3_deletion_count = sum(hrp3_deletion_count)) %>% --> <!-- mutate(total_hrp2_deletion_count = sum(hrp2_deletion_count)) --> <!-- create_dt(previousDeletionCalls_hrp2_hrp3_del_continentSum) --> <!-- ``` --> <!-- #### Field Samples --> <!-- ```{r} --> <!-- previousDeletionCalls_hrp2_del_continentSum_field = previousDeletionCalls %>% --> <!-- filter(IsFieldSample & possiblyHRP2Deleted) %>% --> <!-- group_by(secondaryRegion) %>% --> <!-- count(name = "hrp2_deletion_count") %>% --> <!-- ungroup() %>% --> <!-- mutate(total_hrp2_deletion_count = sum(hrp2_deletion_count)) --> <!-- previousDeletionCalls_hrp3_del_continentSum_field = previousDeletionCalls %>% --> <!-- filter(IsFieldSample & possiblyHRP3Deleted) %>% --> <!-- group_by(secondaryRegion) %>% --> <!-- count(name = "hrp3_deletion_count") %>% --> <!-- ungroup() %>% --> <!-- mutate(total_hrp3_deletion_count = sum(hrp3_deletion_count)) --> <!-- previousDeletionCalls_hrp2_and_hrp3_del_continentSum_field = previousDeletionCalls %>% --> <!-- filter(IsFieldSample & possiblyHRP2Deleted & possiblyHRP3Deleted) %>% --> <!-- group_by(secondaryRegion) %>% --> <!-- count(name = "hrp2_and_hrp3_deletion_count") %>% --> <!-- ungroup() %>% --> <!-- mutate(total_hrp2_and_hrp3_deletion_count = sum(hrp2_and_hrp3_deletion_count)) --> <!-- previousDeletionCalls_hrp2_hrp3_del_continentSum_field = previousDeletionCalls_hrp2_del_continentSum_field %>% --> <!-- full_join(previousDeletionCalls_hrp3_del_continentSum_field) %>% --> <!-- full_join(previousDeletionCalls_hrp2_and_hrp3_del_continentSum_field) %>% --> <!-- ungroup() %>% --> <!-- select(-starts_with("total")) %>% --> <!-- mutate_if(is.numeric,dplyr::coalesce,0) %>% --> <!-- mutate(total_hrp2_and_hrp3_deletion_count = sum(hrp2_and_hrp3_deletion_count)) %>% --> <!-- mutate(total_hrp3_deletion_count = sum(hrp3_deletion_count)) %>% --> <!-- mutate(total_hrp2_deletion_count = sum(hrp2_deletion_count)) --> <!-- create_dt(previousDeletionCalls_hrp2_hrp3_del_continentSum_field) --> <!-- ``` --> ```{r} = previousDeletionCalls %>% filter ("Pattern 2" == HRP3_deletionPattern)= readr:: read_tsv ("samplesCovered.txt" , col_names = "sample" ) %>% filter (sample %!in% samplesToRemove$ sample)cat (coveredSamples$ sample, file = "samplesCovered.txt" , sep = " \n " )= masterTable %>% filter (PreferredSample) %>% left_join (previousDeletionCalls) %>% mutate (uncallable = sample %!in% coveredSamples$ sample) %>% rename (HRP2Call = possiblyHRP2Deleted, HRP3Call = possiblyHRP3Deleted, HRPsCall = hrpCall, Chr11TelemericCall = possiblyChr11Deleted) %>% mutate (HRP2Call = case_when (uncallable ~ "uncallable" , is.na (HRP2Call) ~ "present" , ! is.na (HRP2Call) & HRP2Call ~ "deletion" , ~ "present" ), HRP3Call = case_when (uncallable ~ "uncallable" , is.na (HRP3Call) ~ "present" , ! is.na (HRP3Call) & HRP3Call ~ "deletion" , ~ "present" ), HRPsCall = case_when (uncallable ~ "uncallable" ,is.na (HRPsCall) ~ "pfhrp2+/pfhrp3+" ,~ HRPsCall), Chr11TelemericCall = case_when (uncallable ~ "uncallable" ,is.na (Chr11TelemericCall) ~ "present" , ~ "deleted" %>% left_join (cov %>% select (sample, meanPerBaseCov)) %>% select (- sample, - ExperimentSample,- PreferredSample) %>% rename (sample = BiologicalSample) %>% filter (! is.na (meanPerBaseCov))= masterTable_withDeletionMetaOutput %>% filter (IsFieldSample)write_tsv (masterTable_withDeletionMetaOutput, "Supplemental Table - sample metadata old.tsv" )``` ```{r} = allBasicInfo_filt %>% filter (sample %in% previousDeletionCalls_pattern2$ sample)= allBasicInfo_filt_pat2 %>% filter (` #chrom ` == "Pf3D7_13_v3" ) %>% filter (perBaseCoverageNormRounded > 0 , > 5 )= allBasicInfo_filt_pat2_chr13WithCov %>% group_by (sample) %>% slice_max (end)= allBasicInfo_filt_pat2_chr13WithCov_lastRegion %>% mutate (coreGenomeDeletion = 2853482 - end):: skim (allBasicInfo_filt_pat2_chr13WithCov_lastRegion %>% ungroup () %>% select (sample, coreGenomeDeletion, end, start))``` ```{r} = meta %>% left_join (previousDeletionCalls) %>% rename (hrp3DeletionPattern = HRP3_deletionPattern)= meta[match (allBasicInfo_filt_sp$ sample, meta$ sample), ]= metaSelected %>% filter (possiblyHRP2Deleted)= metaSelected %>% filter (possiblyHRP3Deleted)= metaSelected %>% filter (possiblyHRP2Deleted, possiblyHRP3Deleted)= regions %>% select (name, genomicID)write_tsv (metaSelected, "metaSelected.tab.txt" )write_tsv (regions, "subwindows_regionMeta.tab.txt" )``` ```{r} = previousDeletionCalls %>% filter (possiblyHRP2Deleted)= previousDeletionCalls %>% filter (possiblyChr11Deleted)cat (previousDeletionCalls_possiblyChr11Deleted$ sample, file = "allMetaDeletionCalls_possiblyChr11Deleted_samples.txt" , sep = " \n " )cat (previousDeletionCalls_possiblyHRP2Deleted$ sample, file = "allMetaDeletionCalls_possiblyHRP2Deleted_samples.txt" , sep = " \n " )cat (previousDeletionCalls$ sample, file = "allMetaDeletionCalls_samples.txt" , sep = " \n " )``` ```{r} = readr:: read_tsv ("allMetaDeletionCalls_Hrp3pattern2.tab.txt" )cat (previousDeletionCalls_Hrp3pattern2$ sample, file = "allMetaDeletionCalls_Hrp3pattern2_samples.txt" , sep = " \n " )``` ## Heatmaps ### All deletions ```{r} library (circlize)= colorRamp2 (c (0 , 1 , 2 ), c (heat.colors (3 )))= allBasicInfo_filt_sp_matrownames (allBasicInfo_filt_sp_mat_noLabs) = NULL colnames (allBasicInfo_filt_sp_mat_noLabs) = NULL = metaSelected$ BiologicalSample$ site != "LabIsolate" | is.na (metaSelected$ site)] = "" #RowLabs[metaSelected$country != "Ethiopia"] = "" # rownames(allBasicInfo_filt_sp_mat_noLabs) = RowLabs # regionNames = sort(unique(c(metaSelected$country, metaSelected$region, metaSelected$secondaryRegion))) # regionColors = scheme$hex(length(regionNames)) # names(regionColors) = regionNames = regions[,c ("inGene" , "genomicRegion" , "chrom" )] %>% as.data.frame ()= list ()for (topAnnoName in colnames (topAnnoDf)){= sort (unique (topAnnoDf[[topAnnoName]]))<- iwanthue (seed = rnorm (1 ) * 100 , force_init = TRUE ) if (length (levels) <= 8 ) {#levelsCols = tableau_color_pal()(length(levels)) = palette.colors (palette = "Okabe-Ito" )[c (2 , 3 , 4 , 6 , 7 , 8 , 5 , 9 , 1 )][1 : length (levels)]#levelsCols = sample(scheme$hex(10), length(levels)) else {= scheme$ hex (length (levels))names (levelsCols) = levels= levelsCols# topAnnoColors[["inGene"]] = c("TRUE" = tableau_color_pal()(8)[5], "FALSE" = paste0(tableau_color_pal()(8)[3],"FF")) # topAnnoColors[["homologousRegion"]] = c("other" = paste0(tableau_color_pal()(8)[7], "FF"), "shared" = tableau_color_pal()(8)[8]) # topAnnoColors[["afterHomologousRegion"]] = c("TRUE" = tableau_color_pal()(8)[6], "FALSE" = paste0(tableau_color_pal()(8)[5],"FF")) "inGene" ]] = c ("TRUE" = "#0072B2" , "FALSE" = "#D55E0000" )"genomicRegion" ]][["other" ]] = "#0072B200" = metaSelected[,c ("hrpCall" , "hrp3DeletionPattern" , "isolate" , "country" , "region" , "secondaryRegion" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()= list ()for (rowAnnoName in colnames (rowAnnoDf)){= sort (unique (rowAnnoDf[[rowAnnoName]]))<- iwanthue (seed = rnorm (1 ) * 100 , force_init = TRUE )if (length (levels) <= 9 ) {#levelsCols = tableau_color_pal()(length(levels)) = palette.colors (palette = "Okabe-Ito" )[c (2 , 3 , 4 , 6 , 7 , 8 , 5 , 9 , 1 )][1 : length (levels)]#levelsCols = sample(scheme$hex(10), length(levels)) else {= scheme$ hex (length (levels))names (levelsCols) = levels= levelsCols"region" ]]["Papua New Guinea" ] = "#999999" "region" ]]["South East Asia - West" ] = "#56B4E9" "PerfectChr11Copy" ]] = c ("TRUE" = "#0072B2" , "FALSE" = "#D55E0000" )"hrp3Pattern1" ]] = c ("TRUE" = "#0072B2" , "FALSE" = "#D55E0000" )"HRP3_deletionPattern" ]] = c ("Pattern 1" = "#0072B2" , "Pattern 2" = "#D55E00" )"hrp2" ]] = c ("hrp2+" = "#00C2F9" , "hrp2-" = "#E20134" )"hrpCall" ]] = pfhrpsCallColors"hrpCalls" ]] = pfhrpsCallColors"pfhrp calls" ]] = pfhrpsCallColors# rowAnnoColors[["continent"]] = c("AFRICA" = tableau_color_pal()(8)[1], # "ASIA" = tableau_color_pal()(8)[7], # "OCEANIA" = tableau_color_pal()(8)[2], # "S_AMERICA" = tableau_color_pal()(8)[3]) "continent" ]] = continentColorssave (rowAnnoColors, file = "rowAnnoColors.Rdata" )= 20 = HeatmapAnnotation (df = topAnnoDf,col = topAnnoColors,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" = rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )= Heatmap (cluster_columns = F,col = col_fun,name = "coverage" ,top_annotation = topAnno,left_annotation = sideAnno,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )``` * 1) segmental deletion of chr 13 just centromeric to hrp3 extending to the end of the chromosome, with simultaneous duplication of the chr 11 subtelomeric region, and * 2) deletion of chr 13 starting at various locations centromeric to hrp3 but not involving chr 11 duplication. ```{r} #| fig-column: screen #| fig-width: 30 #| fig-height: 25 draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )``` ```{r} pdf ("allHeatmap.pdf" , useDingbats = F, width = 30 , height = 25 )draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )dev.off ()``` ### HRP2- ```{r} library (circlize)= colorRamp2 (c (0 , 1 , 2 ), c (heat.colors (3 )))= allBasicInfo_filt_sp_mat[rownames (allBasicInfo_filt_sp_mat) %in% metaSelected_hrp2_deleted$ sample, ]= meta[match (rownames (allBasicInfo_filt_sp_mat_selected), meta$ sample), ]= allBasicInfo_filt_sp_mat_selected#rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = NULL colnames (allBasicInfo_filt_sp_mat_selected_noLabs) = NULL = metaReSelected$ BiologicalSample$ site != "LabIsolate" | is.na (metaReSelected$ site)] = "" # rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = RowLabs # regionNames = sort(unique(c(metaReSelected$country, metaReSelected$region, metaReSelected$secondaryRegion))) # regionColors = scheme$hex(length(regionNames)) # names(regionColors) = regionNames = regions[,c ("inGene" , "genomicRegion" , "chrom" )] %>% as.data.frame ()# topAnnoColors = list() # for(topAnnoName in colnames(topAnnoDf)){ # levels = sort(unique(topAnnoDf[[topAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # topAnnoColors[[topAnnoName]] = levelsCols # } # topAnnoColors[["inGene"]] = c("TRUE" = tableau_color_pal()(8)[5], "FALSE" = paste0(tableau_color_pal()(8)[3],"FF")) # topAnnoColors[["homologousRegion"]] = c("other" = paste0(tableau_color_pal()(8)[7], "FF"), "shared" = tableau_color_pal()(8)[8]) # topAnnoColors[["afterHomologousRegion"]] = c("TRUE" = tableau_color_pal()(8)[6], "FALSE" = paste0(tableau_color_pal()(8)[5],"FF")) # # = metaReSelected[,c ("hrpCall" , "hrp3DeletionPattern" , "isolate" , "country" , "region" , "secondaryRegion" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()# # rowAnnoColors = list() # for(rowAnnoName in colnames(rowAnnoDf)){ # levels = sort(unique(rowAnnoDf[[rowAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # rowAnnoColors[[rowAnnoName]] = levelsCols # } # rowAnnoColors[["continent"]] = c("AFRICA" = tableau_color_pal()(8)[1], # "ASIA" = tableau_color_pal()(8)[4], # "OCEANIA" = tableau_color_pal()(8)[2], # "S_AMERICA" = tableau_color_pal()(8)[3]) = 20 = HeatmapAnnotation (df = topAnnoDf,col = topAnnoColors,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" = rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )= Heatmap (cluster_columns = F,col = col_fun,name = "coverage" ,top_annotation = topAnno,left_annotation = sideAnno,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )``` ```{r} #| fig-column: screen #| fig-width: 30 #| fig-height: 15 draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )``` ```{r} cairo_pdf ("allHeatmap_hrp2_deleted.pdf" , width = 30 , height = 15 )draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )dev.off ()``` ### HRP3- ```{r} library (circlize)= colorRamp2 (c (0 , 1 , 2 ), c (heat.colors (3 )))= allBasicInfo_filt_sp_mat[rownames (allBasicInfo_filt_sp_mat) %in% metaSelected_hrp3_deleted$ sample, ]= meta[match (rownames (allBasicInfo_filt_sp_mat_selected), meta$ sample), ]= allBasicInfo_filt_sp_mat_selected#rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = NULL colnames (allBasicInfo_filt_sp_mat_selected_noLabs) = NULL = metaReSelected$ BiologicalSample$ site != "LabIsolate" | is.na (metaReSelected$ site)] = "" # rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = RowLabs # regionNames = sort(unique(c(metaReSelected$country, metaReSelected$region, metaReSelected$secondaryRegion))) # regionColors = scheme$hex(length(regionNames)) # names(regionColors) = regionNames = regions[,c ("inGene" , "genomicRegion" , "chrom" )] %>% as.data.frame ()# topAnnoColors = list() # for(topAnnoName in colnames(topAnnoDf)){ # levels = sort(unique(topAnnoDf[[topAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # topAnnoColors[[topAnnoName]] = levelsCols # } # topAnnoColors[["inGene"]] = c("TRUE" = tableau_color_pal()(8)[5], "FALSE" = paste0(tableau_color_pal()(8)[3],"FF")) # topAnnoColors[["homologousRegion"]] = c("other" = paste0(tableau_color_pal()(8)[7], "FF"), "shared" = tableau_color_pal()(8)[8]) # topAnnoColors[["afterHomologousRegion"]] = c("TRUE" = tableau_color_pal()(8)[6], "FALSE" = paste0(tableau_color_pal()(8)[5],"FF")) # = metaReSelected[,c ("hrpCall" , "hrp3DeletionPattern" , "isolate" , "country" , "region" , "secondaryRegion" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()# rowAnnoColors = list() # for(rowAnnoName in colnames(rowAnnoDf)){ # levels = sort(unique(rowAnnoDf[[rowAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # rowAnnoColors[[rowAnnoName]] = levelsCols # } # rowAnnoColors[["continent"]] = c("AFRICA" = tableau_color_pal()(8)[1], # "ASIA" = tableau_color_pal()(8)[4], # "OCEANIA" = tableau_color_pal()(8)[2], # "S_AMERICA" = tableau_color_pal()(8)[3]) = 20 = HeatmapAnnotation (df = topAnnoDf,col = topAnnoColors,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" = rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )= Heatmap (cluster_columns = F,col = col_fun,name = "coverage" ,top_annotation = topAnno,left_annotation = sideAnno,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )``` ```{r} #| fig-column: screen #| fig-width: 30 #| fig-height: 20 draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )``` ```{r} cairo_pdf ("allHeatmap_hrp3_deleted.pdf" , width = 30 , height = 40 )draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )dev.off ()``` ### pfhrp2-/pfhrp3- ```{r} library (circlize)= colorRamp2 (c (0 , 1 , 2 ), c (heat.colors (3 )))= allBasicInfo_filt_sp_mat[rownames (allBasicInfo_filt_sp_mat) %in% metaSelected_hrp2_and_hrp3_deleted$ sample, ]= meta[match (rownames (allBasicInfo_filt_sp_mat_selected), meta$ sample), ]= allBasicInfo_filt_sp_mat_selected#rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = NULL colnames (allBasicInfo_filt_sp_mat_selected_noLabs) = NULL = metaReSelected$ BiologicalSample$ site != "LabIsolate" | is.na (metaReSelected$ site)] = "" # rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = RowLabs # regionNames = sort(unique(c(metaReSelected$country, metaReSelected$region, metaReSelected$secondaryRegion))) # regionColors = scheme$hex(length(regionNames)) # names(regionColors) = regionNames = regions[,c ("inGene" , "genomicRegion" , "chrom" )] %>% as.data.frame ()# topAnnoColors = list() # for(topAnnoName in colnames(topAnnoDf)){ # levels = sort(unique(topAnnoDf[[topAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # topAnnoColors[[topAnnoName]] = levelsCols # } # topAnnoColors[["inGene"]] = c("TRUE" = tableau_color_pal()(8)[5], "FALSE" = paste0(tableau_color_pal()(8)[3],"FF")) # topAnnoColors[["homologousRegion"]] = c("other" = paste0(tableau_color_pal()(8)[7], "FF"), "shared" = tableau_color_pal()(8)[8]) # topAnnoColors[["afterHomologousRegion"]] = c("TRUE" = tableau_color_pal()(8)[6], "FALSE" = paste0(tableau_color_pal()(8)[5],"FF")) # # = metaReSelected[,c ("hrpCall" , "hrp3DeletionPattern" , "isolate" , "country" , "region" , "secondaryRegion" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()# # rowAnnoColors = list() # for(rowAnnoName in colnames(rowAnnoDf)){ # levels = sort(unique(rowAnnoDf[[rowAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # rowAnnoColors[[rowAnnoName]] = levelsCols # } # rowAnnoColors[["continent"]] = c("AFRICA" = tableau_color_pal()(8)[1], # "ASIA" = tableau_color_pal()(8)[4], # "OCEANIA" = tableau_color_pal()(8)[2], # "S_AMERICA" = tableau_color_pal()(8)[3]) # = 20 = HeatmapAnnotation (df = topAnnoDf,col = topAnnoColors,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" = rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )= Heatmap (cluster_columns = F,col = col_fun,name = "coverage" ,top_annotation = topAnno,left_annotation = sideAnno,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )``` ```{r} #| fig-column: screen #| fig-width: 30 #| fig-height: 20 draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )``` ```{r} cairo_pdf ("allHeatmap_hrp2_and_hrp3_deleted.pdf" , width = 30 , height = 20 )draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )dev.off ()``` ## Heatmaps with removed regions ### Removing regions ```{r} = regions %>% filter ("Pf332" == genomicRegion)= allBasicInfo_filt_sp_mat[,colnames (allBasicInfo_filt_sp_mat) %in% regions_Pf332$ genomicID]= as_tibble (allBasicInfo_filt_sp_mat_Pf332)= allBasicInfo_filt_sp_mat_Pf332_df %>% mutate (sample = rownames (allBasicInfo_filt_sp_mat_Pf332)) %>% gather (genomicID, coverage, 1 : (ncol (.) - 1 ))= allBasicInfo_filt_sp_mat_Pf332_df_long %>% group_by (genomicID) %>% summarise (totalSamps = length (unique (sample)), covered = sum (coverage > 0 )) %>% mutate (coverageRate = covered/ totalSamps)= allBasicInfo_filt_sp_mat_Pf332_df_long_sum %>% filter (coverageRate < 0.90 )= allBasicInfo_filt_sp_mat[,colnames (allBasicInfo_filt_sp_mat) %!in% allBasicInfo_filt_sp_mat_Pf332_df_long_sum_notPass$ genomicID]= regions %>% filter (genomicID %!in% allBasicInfo_filt_sp_mat_Pf332_df_long_sum_notPass$ genomicID)save (allBasicInfo_filt_sp_mat, regions, file = "allBasicInfo_filt_sp_mat.Rdata" )``` ### All deletions ```{r} library (circlize)= colorRamp2 (c (0 , 1 , 2 ), c (heat.colors (3 )))= allBasicInfo_filt_sp_matrownames (allBasicInfo_filt_sp_mat_noLabs) = NULL colnames (allBasicInfo_filt_sp_mat_noLabs) = NULL = metaSelected$ BiologicalSample$ site != "LabIsolate" | is.na (metaSelected$ site)] = "" #RowLabs[metaSelected$country != "Ethiopia"] = "" # rownames(allBasicInfo_filt_sp_mat_noLabs) = RowLabs # regionNames = sort(unique(c(metaSelected$country, metaSelected$region, metaSelected$secondaryRegion))) # regionColors = scheme$hex(length(regionNames)) # names(regionColors) = regionNames = regions[,c ("inGene" , "genomicRegion" , "chrom" )] %>% as.data.frame ()= metaSelected[,c ("hrpCall" , "hrp3DeletionPattern" , "isolate" , "country" , "region" , "secondaryRegion" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()= 20 = HeatmapAnnotation (df = topAnnoDf,col = topAnnoColors,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" = rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )= Heatmap (cluster_columns = F,col = col_fun,name = "coverage" ,top_annotation = topAnno,left_annotation = sideAnno,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )``` * 1) segmental deletion of chr 13 just centromeric to hrp3 extending to the end of the chromosome, with simultaneous duplication of the chr 11 subtelomeric region, and * 2) deletion of chr 13 starting at various locations centromeric to hrp3 but not involving chr 11 duplication. ```{r} #| fig-column: screen #| fig-width: 30 #| fig-height: 25 draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )``` ```{r} pdf ("allHeatmap_filtered.pdf" , useDingbats = F, width = 30 , height = 25 )draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )dev.off ()``` ### HRP2- ```{r} library (circlize)= colorRamp2 (c (0 , 1 , 2 ), c (heat.colors (3 )))= allBasicInfo_filt_sp_mat[rownames (allBasicInfo_filt_sp_mat) %in% metaSelected_hrp2_deleted$ sample, ]= meta[match (rownames (allBasicInfo_filt_sp_mat_selected), meta$ sample), ]= allBasicInfo_filt_sp_mat_selected#rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = NULL colnames (allBasicInfo_filt_sp_mat_selected_noLabs) = NULL = metaReSelected$ BiologicalSample$ site != "LabIsolate" | is.na (metaReSelected$ site)] = "" # rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = RowLabs # regionNames = sort(unique(c(metaReSelected$country, metaReSelected$region, metaReSelected$secondaryRegion))) # regionColors = scheme$hex(length(regionNames)) # names(regionColors) = regionNames = regions[,c ("inGene" , "genomicRegion" , "chrom" )] %>% as.data.frame ()# topAnnoColors = list() # for(topAnnoName in colnames(topAnnoDf)){ # levels = sort(unique(topAnnoDf[[topAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # topAnnoColors[[topAnnoName]] = levelsCols # } # topAnnoColors[["inGene"]] = c("TRUE" = tableau_color_pal()(8)[5], "FALSE" = paste0(tableau_color_pal()(8)[3],"FF")) # topAnnoColors[["homologousRegion"]] = c("other" = paste0(tableau_color_pal()(8)[7], "FF"), "shared" = tableau_color_pal()(8)[8]) # topAnnoColors[["afterHomologousRegion"]] = c("TRUE" = tableau_color_pal()(8)[6], "FALSE" = paste0(tableau_color_pal()(8)[5],"FF")) # # = metaReSelected[,c ("hrpCall" , "hrp3DeletionPattern" , "isolate" , "country" , "region" , "secondaryRegion" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()# # rowAnnoColors = list() # for(rowAnnoName in colnames(rowAnnoDf)){ # levels = sort(unique(rowAnnoDf[[rowAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # rowAnnoColors[[rowAnnoName]] = levelsCols # } # rowAnnoColors[["continent"]] = c("AFRICA" = tableau_color_pal()(8)[1], # "ASIA" = tableau_color_pal()(8)[4], # "OCEANIA" = tableau_color_pal()(8)[2], # "S_AMERICA" = tableau_color_pal()(8)[3]) = 20 = HeatmapAnnotation (df = topAnnoDf,col = topAnnoColors,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" = rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )= Heatmap (cluster_columns = F,col = col_fun,name = "coverage" ,top_annotation = topAnno,left_annotation = sideAnno,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )``` ```{r} #| fig-column: screen #| fig-width: 30 #| fig-height: 15 draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )``` ```{r} pdf ("allHeatmap_hrp2_deleted_filtered.pdf" , useDingbats = F, width = 30 , height = 15 )draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )dev.off ()``` ### HRP3- ```{r} library (circlize)= colorRamp2 (c (0 , 1 , 2 ), c (heat.colors (3 )))= allBasicInfo_filt_sp_mat[rownames (allBasicInfo_filt_sp_mat) %in% metaSelected_hrp3_deleted$ sample, ]= meta[match (rownames (allBasicInfo_filt_sp_mat_selected), meta$ sample), ]= allBasicInfo_filt_sp_mat_selected#rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = NULL colnames (allBasicInfo_filt_sp_mat_selected_noLabs) = NULL = metaReSelected$ BiologicalSample$ site != "LabIsolate" | is.na (metaReSelected$ site)] = "" # rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = RowLabs # regionNames = sort(unique(c(metaReSelected$country, metaReSelected$region, metaReSelected$secondaryRegion))) # regionColors = scheme$hex(length(regionNames)) # names(regionColors) = regionNames = regions[,c ("inGene" , "genomicRegion" , "chrom" )] %>% as.data.frame ()# topAnnoColors = list() # for(topAnnoName in colnames(topAnnoDf)){ # levels = sort(unique(topAnnoDf[[topAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # topAnnoColors[[topAnnoName]] = levelsCols # } # topAnnoColors[["inGene"]] = c("TRUE" = tableau_color_pal()(8)[5], "FALSE" = paste0(tableau_color_pal()(8)[3],"FF")) # topAnnoColors[["homologousRegion"]] = c("other" = paste0(tableau_color_pal()(8)[7], "FF"), "shared" = tableau_color_pal()(8)[8]) # topAnnoColors[["afterHomologousRegion"]] = c("TRUE" = tableau_color_pal()(8)[6], "FALSE" = paste0(tableau_color_pal()(8)[5],"FF")) # = metaReSelected[,c ("hrpCall" , "hrp3DeletionPattern" , "isolate" , "country" , "region" , "secondaryRegion" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()# rowAnnoColors = list() # for(rowAnnoName in colnames(rowAnnoDf)){ # levels = sort(unique(rowAnnoDf[[rowAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # rowAnnoColors[[rowAnnoName]] = levelsCols # } # rowAnnoColors[["continent"]] = c("AFRICA" = tableau_color_pal()(8)[1], # "ASIA" = tableau_color_pal()(8)[4], # "OCEANIA" = tableau_color_pal()(8)[2], # "S_AMERICA" = tableau_color_pal()(8)[3]) = 20 = HeatmapAnnotation (df = topAnnoDf,col = topAnnoColors,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" = rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )= Heatmap (cluster_columns = F,col = col_fun,name = "coverage" ,top_annotation = topAnno,left_annotation = sideAnno,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )``` ```{r} #| fig-column: screen #| fig-width: 30 #| fig-height: 20 draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )``` ```{r} pdf ("allHeatmap_hrp3_deleted_filtered.pdf" , useDingbats = F, width = 30 , height = 20 )draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )dev.off ()``` ### pfhrp2-/pfhrp3- ```{r} library (circlize)= colorRamp2 (c (0 , 1 , 2 ), c (heat.colors (3 )))= allBasicInfo_filt_sp_mat[rownames (allBasicInfo_filt_sp_mat) %in% metaSelected_hrp2_and_hrp3_deleted$ sample, ]= meta[match (rownames (allBasicInfo_filt_sp_mat_selected), meta$ sample), ]= allBasicInfo_filt_sp_mat_selected#rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = NULL colnames (allBasicInfo_filt_sp_mat_selected_noLabs) = NULL = metaReSelected$ BiologicalSample$ site != "LabIsolate" | is.na (metaReSelected$ site)] = "" # rownames(allBasicInfo_filt_sp_mat_selected_noLabs) = RowLabs # regionNames = sort(unique(c(metaReSelected$country, metaReSelected$region, metaReSelected$secondaryRegion))) # regionColors = scheme$hex(length(regionNames)) # names(regionColors) = regionNames = regions[,c ("inGene" , "genomicRegion" , "chrom" )] %>% as.data.frame ()# topAnnoColors = list() # for(topAnnoName in colnames(topAnnoDf)){ # levels = sort(unique(topAnnoDf[[topAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # topAnnoColors[[topAnnoName]] = levelsCols # } # topAnnoColors[["inGene"]] = c("TRUE" = tableau_color_pal()(8)[5], "FALSE" = paste0(tableau_color_pal()(8)[3],"FF")) # topAnnoColors[["homologousRegion"]] = c("other" = paste0(tableau_color_pal()(8)[7], "FF"), "shared" = tableau_color_pal()(8)[8]) # topAnnoColors[["afterHomologousRegion"]] = c("TRUE" = tableau_color_pal()(8)[6], "FALSE" = paste0(tableau_color_pal()(8)[5],"FF")) # # = metaReSelected[,c ("hrpCall" , "hrp3DeletionPattern" , "isolate" , "country" , "region" , "secondaryRegion" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()# # rowAnnoColors = list() # for(rowAnnoName in colnames(rowAnnoDf)){ # levels = sort(unique(rowAnnoDf[[rowAnnoName]])) # scheme <- iwanthue(seed = rnorm(1) * 100, force_init = TRUE) # if(length(levels) <= 10){ # levelsCols = tableau_color_pal()(length(levels)) # #levelsCols = sample(scheme$hex(10), length(levels)) # }else{ # levelsCols = scheme$hex(length(levels)) # } # names(levelsCols) = levels # rowAnnoColors[[rowAnnoName]] = levelsCols # } # rowAnnoColors[["continent"]] = c("AFRICA" = tableau_color_pal()(8)[1], # "ASIA" = tableau_color_pal()(8)[4], # "OCEANIA" = tableau_color_pal()(8)[2], # "S_AMERICA" = tableau_color_pal()(8)[3]) # = 20 = HeatmapAnnotation (df = topAnnoDf,col = topAnnoColors,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" = rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )= Heatmap (cluster_columns = F,col = col_fun,name = "coverage" ,top_annotation = topAnno,left_annotation = sideAnno,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )``` ```{r} #| fig-column: screen #| fig-width: 30 #| fig-height: 20 draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )``` ```{r} pdf ("allHeatmap_hrp2_and_hrp3_deleted_filtered.pdf" , useDingbats = F, width = 30 , height = 20 )draw (allCoverageHeatMap, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "left" , annotation_legend_side = "left" )dev.off ()``` <!-- ## By chromosome --> <!-- ### All --> <!-- ```{r} --> <!-- library(circlize) --> <!-- col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3))) --> <!-- allBasicInfo_filt_sp_mat_noLabs = allBasicInfo_filt_sp_mat --> <!-- rownames(allBasicInfo_filt_sp_mat_noLabs) = NULL --> <!-- colnames(allBasicInfo_filt_sp_mat_noLabs) = NULL --> <!-- rowAnnoDf = metaSelected[,c("hrpCall", "hrp3DeletionPattern", "isolate", "country", "region", "secondaryRegion")] %>% rename(continent = secondaryRegion) %>% as.data.frame() --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr08 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_08_", colnames(allBasicInfo_filt_sp_mat))] #08: 1290365 - 1387982 --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr11 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_11_", colnames(allBasicInfo_filt_sp_mat))] #11: 1897157 - 2003328 --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr13 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_13_", colnames(allBasicInfo_filt_sp_mat))] #13: 2769916 - 2844777 --> <!-- #08: 1290365 - 1387982 --> <!-- #11: 1897157 - 2003328 --> <!-- #13: 2769916 - 2844777 --> <!-- # topAnnoDf = regions[,c("inGene", "genomicRegion", "chrom")] %>% as.data.frame() --> <!-- topAnnoDf = regions[,c("inGene", "genomicRegion", "chrom")] %>% as.data.frame() --> <!-- topAnnoDf_chr08 = topAnnoDf %>% filter(grepl("_08_",chrom)) %>% select(-chrom) --> <!-- topAnnoDf_chr11 = topAnnoDf %>% filter(grepl("_11_",chrom)) %>% select(-chrom) --> <!-- topAnnoDf_chr13 = topAnnoDf %>% filter(grepl("_13_",chrom)) %>% select(-chrom) --> <!-- annotationTextSize = 20 --> <!-- topAnno_chr08 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr08, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left" --> <!-- ) --> <!-- topAnno_chr11 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr11, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- show_annotation_name = F --> <!-- ) --> <!-- topAnno_chr13 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr13, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- show_annotation_name = F --> <!-- ) --> <!-- sideAnno = rowAnnotation( --> <!-- df = rowAnnoDf, --> <!-- col = rowAnnoColors, --> <!-- gp = gpar(col = "grey10"), --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ) --> <!-- ) --> <!-- library(dendsort) --> <!-- clustering = --> <!-- # row_dend = dendsort(hclust(dist(allBasicInfo_filt_sp_mat_noLabs, method = "euclidean"), method = "complete")) --> <!-- # row_dend$method = "euclidean"; row_dend$dist.method = "complete" --> <!-- # col_dend = dendsort(hclust(dist(t(allBasicInfo_filt_sp_mat_noLabs), method = "euclidean"), method = "complete")) --> <!-- # col_dend$method = "euclidean"; col_dend$dist.method = "complete" --> <!-- row_dend = dendsort(hclust(dist(allBasicInfo_filt_sp_mat_noLabs))) --> <!-- allCoverageHeatMap_chr08 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr08, --> <!-- cluster_columns = F, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr08, --> <!-- left_annotation = sideAnno, --> <!-- cluster_rows = row_dend, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr08", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- allCoverageHeatMap_chr11 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr11, --> <!-- cluster_columns = F, --> <!-- cluster_rows = row_dend, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr11, --> <!-- # left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr11", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- allCoverageHeatMap_chr13 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr13, --> <!-- cluster_columns = F, --> <!-- cluster_rows = row_dend, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr13, --> <!-- # left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr13", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- hms_list = allCoverageHeatMap_chr08 + allCoverageHeatMap_chr11 + allCoverageHeatMap_chr13 --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen --> <!-- #| fig-width: 30 --> <!-- #| fig-height: 20 --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- ``` --> <!-- ```{r} --> <!-- pdf("allHeatmap_byChrom_filtered.pdf", useDingbats = F, width = 30, height = 20) --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- dev.off() --> <!-- ``` --> <!-- #### Just chr11 and chr13 --> <!-- ```{r} --> <!-- rowAnnoDf = metaSelected[,c("hrp3DeletionPattern", "secondaryRegion", "hrpCall", "possiblyChr11Deleted")] %>% rename(continent = secondaryRegion --> <!-- # , --> <!-- # hrp2 = possiblyHRP2Deleted --> <!-- ) %>% --> <!-- # mutate(hrp2 = ifelse(hrp2, "hrp2-", "hrp2+")) %>% --> <!-- mutate(hrp3DeletionPattern = case_when( --> <!-- "Pattern 1" == hrp3DeletionPattern ~ "11++/13-", --> <!-- "Pattern 2" == hrp3DeletionPattern ~ "11/13-", --> <!-- possiblyChr11Deleted ~ "11-/13" --> <!-- )) %>% --> <!-- select(-possiblyChr11Deleted) %>% --> <!-- rename(DeletionPattern = hrp3DeletionPattern) %>% --> <!-- as.data.frame() --> <!-- rowAnnoColorsMod = rowAnnoColors --> <!-- rowAnnoColorsMod[["hrp3DeletionPattern"]] = c("11++/13-" = "#8400CD", "1113-" = "#9F0162", "11-/13" = "#FF6E3A") --> <!-- rowAnnoColorsMod[["DeletionPattern"]] = c("11++/13-" = "#8400CD", "11/13-" = "#9F0162", "11-/13" = "#FF6E3A") --> <!-- rowAnnoDf_sorted = rowAnnoDf %>% --> <!-- as_tibble() %>% --> <!-- mutate(rowID = row_number()) %>% --> <!-- arrange() --> <!-- sideAnno = rowAnnotation( --> <!-- df = rowAnnoDf, --> <!-- col = rowAnnoColorsMod, --> <!-- gp = gpar(col = "grey10"), --> <!-- simple_anno_size = unit(1.5, "cm"), --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ) --> <!-- ) --> <!-- topAnno_chr11 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr11, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- show_annotation_name = T --> <!-- ) --> <!-- allCoverageHeatMap_chr11 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr11, --> <!-- cluster_columns = F, --> <!-- cluster_rows = row_dend, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr11, --> <!-- left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr11", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- allCoverageHeatMap_chr13 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr13, --> <!-- cluster_columns = F, --> <!-- cluster_rows = row_dend, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr13, --> <!-- # left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr13", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- hms_list = allCoverageHeatMap_chr11 + allCoverageHeatMap_chr13 --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen --> <!-- #| fig-width: 30 --> <!-- #| fig-height: 20 --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- ``` --> <!-- ```{r} --> <!-- pdf("allHeatmap_byChrom_filtered_chr11_chr13_leftLegend.pdf", useDingbats = F, width = 30, height = 20) --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- dev.off() --> <!-- #pdf("allHeatmap_byChrom_filtered_chr11_chr13.pdf", useDingbats = F, width = 30, height = 20) --> <!-- pdf("allHeatmap_byChrom_filtered_chr11_chr13.pdf", useDingbats = F, width = 16, height = 12) --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", ht_gap = unit(2, "cm")) --> <!-- dev.off() --> <!-- ``` --> <!-- ##### re-sorted --> <!-- ```{r} --> <!-- hrp3_pat2_sample_deletion_meta = readr::read_tsv("hrp3_pat2_sample_deletion_meta.tsv") --> <!-- hrp3_pat2_sample_deletion_meta_transition_chr5 = hrp3_pat2_sample_deletion_meta %>% --> <!-- filter(`Transposition With Chr5`) --> <!-- hrp3_pat2_sample_deletion_meta_tare1_onchr13 = hrp3_pat2_sample_deletion_meta %>% --> <!-- filter(`TARE1 On Chr13`) --> <!-- finalHRPII_HRPIII_windows_investTelemeraseHealing_chr11_full_out_step50_windowSize100_tare1_samples = readr::read_tsv("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr11_full_out_step50_windowSize100/finalHRPII_HRPIII_windows_investTelemeraseHealing_chr11_full_out_step50_windowSize100_tare1_samples.txt", col_names = c("sample")) --> <!-- finalHRPII_HRPIII_windows_investTelemeraseHealing_chr11_full_out_step50_windowSize100_tare1_samplesMeta = readr::read_tsv("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr11_full_out_step50_windowSize100_rowAnno.tsv") %>% --> <!-- filter(`TARE1 On Chr11`) --> <!-- rowAnnoDf = metaSelected[,c("sample", "hrp3DeletionPattern", "secondaryRegion", "hrpCall", "possiblyChr11Deleted")] %>% rename(continent = secondaryRegion --> <!-- # , --> <!-- # hrp2 = possiblyHRP2Deleted --> <!-- ) %>% --> <!-- # mutate(hrp2 = ifelse(hrp2, "hrp2-", "hrp2+")) %>% --> <!-- mutate(hrp3DeletionPattern = case_when( --> <!-- "Pattern 1" == hrp3DeletionPattern ~ "11++/13-", --> <!-- "Pattern 2" == hrp3DeletionPattern ~ "11/13-", --> <!-- possiblyChr11Deleted ~ "11-/13" --> <!-- )) %>% --> <!-- mutate(hrp3DeletionPattern = ifelse(sample %in% hrp3_pat2_sample_deletion_meta_tare1_onchr13$sample, "11/13-TARE1", hrp3DeletionPattern) ) %>% --> <!-- mutate(hrp3DeletionPattern = ifelse(sample %in% hrp3_pat2_sample_deletion_meta_transition_chr5$sample, "5++/13-", hrp3DeletionPattern) ) %>% --> <!-- mutate(hrp3DeletionPattern = ifelse(sample %in% finalHRPII_HRPIII_windows_investTelemeraseHealing_chr11_full_out_step50_windowSize100_tare1_samplesMeta$sample, "11-TARE1/13", hrp3DeletionPattern) ) %>% --> <!-- select(-possiblyChr11Deleted, -sample) %>% --> <!-- rename(DeletionPattern = hrp3DeletionPattern) %>% --> <!-- as.data.frame() --> <!-- rowAnnoColorsMod = rowAnnoColors --> <!-- rowAnnoColorsMod[["hrp3DeletionPattern"]] = c( --> <!-- "11++/13-" = "#8400CD", --> <!-- "11/13-TARE1" = "#003C86", --> <!-- "11/13-" = "#008DF9", --> <!-- "11-/13" = "#9F0162", --> <!-- "11-TARE1/13" = "#EF0096", --> <!-- "5++/13-" = "#FF6E3A" --> <!-- ) --> <!-- rowAnnoColorsMod[["DeletionPattern"]] = c( --> <!-- "11++/13-" = "#8400CD", --> <!-- "11/13-TARE1" = "#003C86", --> <!-- "11/13-" = "#008DF9", --> <!-- "11-/13" = "#9F0162", --> <!-- "11-TARE1/13" = "#EF0096", --> <!-- "5++/13-" = "#FF6E3A" --> <!-- ) --> <!-- rowAnnoColorsMod[["Coverage Pattern"]] = rowAnnoColorsMod[["DeletionPattern"]] --> <!-- rowAnnoColorsMod[["Pattern"]] = rowAnnoColorsMod[["DeletionPattern"]] --> <!-- rowAnnoColorsMod[["hrp Call"]] = rowAnnoColorsMod[["hrpCall"]] --> <!-- rowAnnoColorsMod[["pfhrps Call"]] = rowAnnoColorsMod[["hrpCall"]] --> <!-- topAnnoColors[["Genomic Region"]] = topAnnoColors[["genomicRegion"]] --> <!-- topAnnoColors[["Intragenic"]] = topAnnoColors[["inGene"]] --> <!-- topAnnoColors_mod = topAnnoColors --> <!-- topAnnoColors_mod[["Genes"]] = c("other" = "#56B4E9", hrp = "#009E73", Pf332 = "#D55E00", rRNA = "#FF5AAF", "pfmdr1" = "#7CFFFA") --> <!-- topAnnoColors_mod[["Genes"]] = c("other" = "#56B4E9", hrp = "#8400CD", Pf332 = "#D55E00", rRNA = "#FF5AAF", "pfmdr1" = "#7CFFFA") --> <!-- topAnnoColors_mod[["Genomic Region"]] = c("Chr11/13 Duplicated Region" = "#0072B2", "Subtelomere" = "#E69F00", "Chr5 Duplication" = "#EF0096") --> <!-- #"TARE1 Chr5" = "#F60239", "Transition with 13" = "#F60239") --> <!-- rowAnnoDf_sorted = rowAnnoDf %>% --> <!-- as_tibble() %>% --> <!-- mutate(rowID = row_number()) %>% --> <!-- mutate(DeletionPattern = factor(DeletionPattern, levels = c("11++/13-", "5++/13-", "11/13-TARE1", "11/13-", "11-TARE1/13", "11-/13"))) %>% --> <!-- arrange(DeletionPattern, continent, hrpCall) %>% --> <!-- rename("Pattern" = DeletionPattern, --> <!-- "pfhrps Call" = hrpCall) --> <!-- rowAnnoDf_sortedDf = rowAnnoDf_sorted %>% --> <!-- select(-rowID) %>% --> <!-- as.data.frame() --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr11_sorted = allBasicInfo_filt_sp_mat_noLabs_chr11[rowAnnoDf_sorted$rowID,] --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr13_sorted = allBasicInfo_filt_sp_mat_noLabs_chr13[rowAnnoDf_sorted$rowID,] --> <!-- sideAnno = rowAnnotation( --> <!-- df = rowAnnoDf_sortedDf, --> <!-- col = rowAnnoColorsMod, --> <!-- gp = gpar(col = "grey10"), --> <!-- simple_anno_size = unit(1.5, "cm"), --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- gap = unit(0.1, "cm") --> <!-- ) --> <!-- topAnnoDf_chr11_mod = topAnnoDf_chr11 %>% --> <!-- rename("Genomic Region" = genomicRegion, "Genes" = "inGene") %>% --> <!-- mutate(Genes = ifelse(Genes, "other", NA)) %>% --> <!-- mutate(Genes = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "rRNA"), `Genomic Region`, Genes)) %>% --> <!-- mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "After Duplicated Region"), "Subtelomere", `Genomic Region`))%>% --> <!-- mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("rRNA", "Duplicated Region"), "Chr11/13 Duplicated Region", `Genomic Region`))%>% --> <!-- mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("other"), NA, `Genomic Region`)) --> <!-- topAnnoDf_chr13_mod = topAnnoDf_chr13 %>% --> <!-- rename("Genomic Region" = genomicRegion, "Genes" = "inGene")%>% --> <!-- mutate(Genes = ifelse(Genes, "other", NA)) %>% --> <!-- mutate(Genes = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "rRNA"), `Genomic Region`, Genes)) %>% --> <!-- mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("hrp", "Pf332", "After Duplicated Region"), "Subtelomere", `Genomic Region`))%>% --> <!-- mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("rRNA", "Duplicated Region"), "Chr11/13 Duplicated Region", `Genomic Region`)) %>% --> <!-- mutate(`Genomic Region` = ifelse(`Genomic Region` %in% c("other"), NA, `Genomic Region`)) --> <!-- topAnno_chr11 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr11_mod, --> <!-- col = topAnnoColors_mod, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- show_annotation_name = T, --> <!-- na_col = "#FFFFFF00", --> <!-- gap = unit(0.1, "cm") --> <!-- ) --> <!-- topAnno_chr13 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr13_mod, --> <!-- col = topAnnoColors_mod, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- show_annotation_name = F, --> <!-- na_col = "#FFFFFF00", --> <!-- gap = unit(0.1, "cm") --> <!-- ) --> <!-- allCoverageHeatMap_chr11 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr11_sorted, --> <!-- cluster_columns = F, --> <!-- cluster_rows = F, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr11, --> <!-- left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"), --> <!-- title = "coverage", at = c(0, 0.5, 1, 1.5, 2), --> <!-- labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x") --> <!-- ), --> <!-- column_title = "Chr11", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- allCoverageHeatMap_chr13 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr13_sorted, --> <!-- cluster_columns = F, --> <!-- cluster_rows = F, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr13, --> <!-- # left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold", --> <!-- title = "coverage", at = c(0, 0.5, 1, 1.5, 2), --> <!-- labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")) --> <!-- ), --> <!-- column_title = "Chr13", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- ``` --> <!-- ```{r} --> <!-- hms_list = allCoverageHeatMap_chr11 + allCoverageHeatMap_chr13 --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen --> <!-- #| fig-width: 30 --> <!-- #| fig-height: 20 --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- ``` --> <!-- ```{r} --> <!-- pdf("allHeatmap_byChrom_filtered_chr11_chr13_leftLegend_sorted.pdf", useDingbats = F, width = 30, height = 20) --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- dev.off() --> <!-- ``` --> <!-- ```{r} --> <!-- #pdf("allHeatmap_byChrom_filtered_chr11_chr13.pdf", useDingbats = F, width = 30, height = 20) --> <!-- pdf("allHeatmap_byChrom_filtered_chr11_chr13_sorted.pdf", useDingbats = F, width = 16, height = 12) --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", --> <!-- ht_gap = unit(2, "cm"), padding = unit(c(5.5 *5*2, 5.5, 5.5, 5.5), "points")) --> <!-- dev.off() --> <!-- ``` --> <!-- ```{r} --> <!-- cov = readr::read_tsv("../../../../allSRAData/reProcess_2021_11_19/coverage/data/allCov_summaryStats.tab.txt.gz") --> <!-- allMetaDeletionCalls_Hrp3pattern2_samples = readr::read_tsv("/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_Hrp3pattern2_samples.txt", col_names = "sample") --> <!-- allReports_chr5 = readr::read_tsv("stableWindows_05_regionOfInterestNarrow_step150_windowSize200//reports/allBasicInfo.tab.txt.gz") %>% --> <!-- filter(start >= 944389) %>% --> <!-- #filter(sample %in% allMetaDeletionCalls_Hrp3pattern2_samples$sample) %>% --> <!-- filter(sample %in% rownames(allBasicInfo_filt_sp_mat)) %>% --> <!-- mutate(inGene = !is.na(extraField0) ) %>% --> <!-- left_join(cov) %>% --> <!-- mutate(medianCov = median(perBaseCoverage), --> <!-- meanCov = mean(perBaseCoverage)) %>% --> <!-- mutate(perBaseCoverageNorm = ifelse(inGene, perBaseCoverage/medianPerBaseCov_inGenes, perBaseCoverage/medianPerBaseCov_notInGenes)) %>% --> <!-- mutate(perBaseCoverageNormRounded = ifelse(perBaseCoverageNorm >0.10 & perBaseCoverageNorm <= 1, 1, perBaseCoverageNorm)) %>% --> <!-- mutate(perBaseCoverageNormRounded = round(perBaseCoverageNormRounded)) --> <!-- allReports_chr5_sp = allReports_chr5 %>% --> <!-- select(sample, name, perBaseCoverageNormRounded) %>% --> <!-- spread(name, perBaseCoverageNormRounded) --> <!-- allReports_chr5_sp_mat = as.matrix(allReports_chr5_sp[,2:ncol(allReports_chr5_sp)]) --> <!-- rownames(allReports_chr5_sp_mat) = allReports_chr5_sp$sample --> <!-- allReports_chr5_sp_mat = allReports_chr5_sp_mat[match(rownames(allBasicInfo_filt_sp_mat)[rowAnnoDf_sorted$rowID], rownames(allReports_chr5_sp_mat)), ] --> <!-- allReports_chr5_sp_mat[allReports_chr5_sp_mat >2] = 2 --> <!-- topAnno_chr05 = allReports_chr5 %>% --> <!-- filter(sample == .$sample[1]) %>% --> <!-- select(name, extraField0) %>% --> <!-- rename(`Gene Description` = extraField0) %>% --> <!-- mutate(`Gene Description` = gsub(".*description=", "", `Gene Description`))%>% --> <!-- mutate(`Gene Description` = gsub("\\].*", "", `Gene Description`)) %>% --> <!-- mutate(`Gene Description` = ifelse(grepl("PHIST", `Gene Description`), gsub("\\).*", "", gsub(".*PHIST", "PHIST", `Gene Description`)), `Gene Description`)) --> <!-- transitionPoint = allReports_chr5 %>% --> <!-- filter(sample == .$sample[1]) %>% --> <!-- mutate(diffFromTransition = abs(979203 - start)) %>% --> <!-- mutate(`Transition Point` = ifelse(diffFromTransition <=100, TRUE, NA)) %>% --> <!-- select(name, `Transition Point`) --> <!-- stableWindows_05_regionOfInterestNarrow_step150_windowSize200_sampleRegion = readr::read_tsv("stableWindows_05_regionOfInterestNarrow_step150_windowSize200/stableWindows_05_regionOfInterestNarrow_step150_windowSize200_sampleRegion.txt", col_names = c("sample", "region")) %>% --> <!-- separate(region, into = c("chrom", "start", "end"), sep = "-", convert = T, remove = F) --> <!-- stableWindows_05_regionOfInterestNarrow_step150_windowSize200_sampleRegion_count = stableWindows_05_regionOfInterestNarrow_step150_windowSize200_sampleRegion %>% --> <!-- group_by(sample) %>% --> <!-- slice_min(order_by = start) %>% --> <!-- group_by(region) %>% --> <!-- count(name = "TARE1 Present Chr5 Count") --> <!-- topAnno_chr05 = topAnno_chr05 %>% --> <!-- left_join( --> <!-- stableWindows_05_regionOfInterestNarrow_step150_windowSize200_sampleRegion_count %>% --> <!-- rename(name = region) --> <!-- ) %>% --> <!-- left_join(transitionPoint) --> <!-- topAnno_chr05Df = topAnno_chr05 %>% --> <!-- separate(name, into = c("chrom", "start", "end"), remove = F, convert = T, sep = "-") %>% --> <!-- mutate(`Genomic Region` = ifelse(start >= 952474 & end <= 979431, "Chr5 Duplication", NA)) %>% --> <!-- select(-name, -chrom, -start, -end) %>% --> <!-- as.data.frame() --> <!-- topAnno_chr05Colors = createColorListFromDf(topAnno_chr05Df) --> <!-- # 05: 944389 - 988747 --> <!-- topAnno_chr05Df_mod = topAnno_chr05Df %>% --> <!-- # select(`Gene Description`, `TARE1 Present Chr5 Count`, `Transition Point`) %>% --> <!-- select(`Gene Description`, `Genomic Region`) %>% --> <!-- mutate(Genes = ifelse(is.na(`Gene Description`), NA, "other")) %>% --> <!-- mutate(Genes = ifelse(`Gene Description` == "multidrug resistance protein 1", "pfmdr1", Genes)) %>% --> <!-- #mutate(`Genomic Region` = NA) %>% --> <!-- # mutate(`Genomic Region` = case_when(22 == `TARE1 Present Chr5 Count` ~ "TARE1 Chr5", --> <!-- # `Transition Point` ~ "Transition with 13", --> <!-- # T ~ `Genomic Region` --> <!-- # )) %>% --> <!-- select(`Genes`, `Genomic Region`) --> <!-- # rowAnno = tibble(metaReSelected[,c("sample","country", "secondaryRegion")]) %>% --> <!-- # rename(continent = secondaryRegion) %>% --> <!-- # mutate(`TARE1 On Chr13` = sample %in% hrp3Pat2_samplesWithTare1$sample, --> <!-- # `Transposition With Chr5` = sample %in% samplesWithTransitionToChr5$sample, --> <!-- # `TARE1 On Chr5` = sample %in% stableWindows_05_regionOfInterestNarrow_step150_windowSize200_sampleRegion$sample) --> <!-- # rowAnno = tibble(metaReSelected[,c("sample","country")]) %>% --> <!-- # mutate(`TARE1 On Chr13` = sample %in% hrp3Pat2_samplesWithTare1$sample, --> <!-- # `Transposition With Chr5` = sample %in% samplesWithTransitionToChr5$sample, --> <!-- # `TARE1 On Chr5` = sample %in% stableWindows_05_regionOfInterestNarrow_step150_windowSize200_sampleRegion$sample) --> <!-- # --> <!-- # rowAnnoDf = rowAnno %>% --> <!-- # select(-sample) %>% --> <!-- # as.data.frame() --> <!-- # rowAnnoColors = createColorListFromDf(rowAnnoDf) --> <!-- # sideAnno = rowAnnotation( --> <!-- # df = rowAnnoDf, --> <!-- # col = rowAnnoColors, --> <!-- # gp = gpar(col = "grey10"), --> <!-- # annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- # annotation_legend_param = list( --> <!-- # labels_gp = gpar(fontsize = annotationTextSize), --> <!-- # title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- # ), --> <!-- # gap = unit(0.1, "cm") --> <!-- # ) --> <!-- topAnno_chr05HM = HeatmapAnnotation( --> <!-- df = topAnno_chr05Df_mod, --> <!-- col = topAnnoColors_mod, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- na_col = c("#99999900"), --> <!-- gap = unit(0.1, "cm"), --> <!-- show_annotation_name = F --> <!-- ) --> <!-- library(circlize) --> <!-- col_fun = colorRamp2(c(0, max(allReports_chr5_sp_mat)/2, max(allReports_chr5_sp_mat)), c(heat.colors(3))) --> <!-- allReports_chr5_sp_mat_nolab = allReports_chr5_sp_mat --> <!-- colnames(allReports_chr5_sp_mat_nolab) = NULL --> <!-- rownames(allReports_chr5_sp_mat_nolab) = NULL --> <!-- allReports_chr5_sp_mat_hm = Heatmap( --> <!-- allReports_chr5_sp_mat_nolab, --> <!-- cluster_columns = F, --> <!-- cluster_rows = F, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr05HM, --> <!-- #right_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold", --> <!-- title = "coverage", at = c(0, 0.5, 1, 1.5, 2), --> <!-- labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")) --> <!-- ), --> <!-- column_title = "Chr05", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- ``` --> <!-- ```{r} --> <!-- #05: 944389 - 988747 --> <!-- #11: 1897157 - 2003328 --> <!-- #13: 2769916 - 2844777 --> <!-- hms_list = allCoverageHeatMap_chr11 + allCoverageHeatMap_chr13 + allReports_chr5_sp_mat_hm --> <!-- pdf("allHeatmap_byChrom_filtered_chr11_chr13_chr5_sorted.pdf", useDingbats = F, width = 16, height = 12) --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", --> <!-- ht_gap = unit(2, "cm"), --> <!-- #padding = unit(c(5.5 *5*2, 5.5, 5.5, 5.5), "points") --> <!-- ) --> <!-- dev.off() --> <!-- ``` --> <!-- ### Hrp2- --> <!-- ```{r} --> <!-- allBasicInfo_filt_sp_mat_selected = allBasicInfo_filt_sp_mat[rownames(allBasicInfo_filt_sp_mat) %in% metaSelected_hrp2_deleted$sample, ] --> <!-- metaReSelected = meta[match(rownames(allBasicInfo_filt_sp_mat_selected), meta$sample), ] --> <!-- library(circlize) --> <!-- col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3))) --> <!-- allBasicInfo_filt_sp_mat_noLabs = allBasicInfo_filt_sp_mat_selected --> <!-- rownames(allBasicInfo_filt_sp_mat_noLabs) = NULL --> <!-- colnames(allBasicInfo_filt_sp_mat_noLabs) = NULL --> <!-- rowAnnoDf = metaReSelected[,c("hrpCall", "hrp3DeletionPattern", "isolate", "country", "region", "secondaryRegion")] %>% rename(continent = secondaryRegion) %>% as.data.frame() --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr08 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_08_", colnames(allBasicInfo_filt_sp_mat))] --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr11 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_11_", colnames(allBasicInfo_filt_sp_mat))] --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr13 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_13_", colnames(allBasicInfo_filt_sp_mat))] --> <!-- # topAnnoDf = regions[,c("inGene", "genomicRegion", "chrom")] %>% as.data.frame() --> <!-- topAnnoDf = regions[,c("inGene", "genomicRegion", "chrom")] %>% as.data.frame() --> <!-- topAnnoDf_chr08 = topAnnoDf %>% filter(grepl("_08_",chrom)) %>% select(-chrom) --> <!-- topAnnoDf_chr11 = topAnnoDf %>% filter(grepl("_11_",chrom)) %>% select(-chrom) --> <!-- topAnnoDf_chr13 = topAnnoDf %>% filter(grepl("_13_",chrom)) %>% select(-chrom) --> <!-- annotationTextSize = 20 --> <!-- topAnno_chr08 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr08, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left" --> <!-- ) --> <!-- topAnno_chr11 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr11, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- show_annotation_name = F --> <!-- ) --> <!-- topAnno_chr13 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr13, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- show_annotation_name = F --> <!-- ) --> <!-- sideAnno = rowAnnotation( --> <!-- df = rowAnnoDf, --> <!-- col = rowAnnoColors, --> <!-- gp = gpar(col = "grey10"), --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ) --> <!-- ) --> <!-- library(dendsort) --> <!-- clustering = --> <!-- # row_dend = dendsort(hclust(dist(allBasicInfo_filt_sp_mat_noLabs, method = "euclidean"), method = "complete")) --> <!-- # row_dend$method = "euclidean"; row_dend$dist.method = "complete" --> <!-- # col_dend = dendsort(hclust(dist(t(allBasicInfo_filt_sp_mat_noLabs), method = "euclidean"), method = "complete")) --> <!-- # col_dend$method = "euclidean"; col_dend$dist.method = "complete" --> <!-- row_dend = dendsort(hclust(dist(allBasicInfo_filt_sp_mat_noLabs))) --> <!-- allCoverageHeatMap_chr08 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr08, --> <!-- cluster_columns = F, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr08, --> <!-- left_annotation = sideAnno, --> <!-- cluster_rows = row_dend, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr08", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- allCoverageHeatMap_chr11 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr11, --> <!-- cluster_columns = F, --> <!-- cluster_rows = row_dend, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr11, --> <!-- # left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr11", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- allCoverageHeatMap_chr13 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr13, --> <!-- cluster_columns = F, --> <!-- cluster_rows = row_dend, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr13, --> <!-- # left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr13", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- hms_list = allCoverageHeatMap_chr08 + allCoverageHeatMap_chr11 + allCoverageHeatMap_chr13 --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen --> <!-- #| fig-width: 30 --> <!-- #| fig-height: 15 --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- ``` --> <!-- ```{r} --> <!-- pdf("allHeatmap_byChrom_hrp2_deleted_filtered.pdf", useDingbats = F, width = 30, height = 15) --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- dev.off() --> <!-- ``` --> <!-- ### Hrp3- --> <!-- ```{r} --> <!-- allBasicInfo_filt_sp_mat_selected = allBasicInfo_filt_sp_mat[rownames(allBasicInfo_filt_sp_mat) %in% metaSelected_hrp3_deleted$sample, ] --> <!-- metaReSelected = meta[match(rownames(allBasicInfo_filt_sp_mat_selected), meta$sample), ] --> <!-- library(circlize) --> <!-- col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3))) --> <!-- allBasicInfo_filt_sp_mat_noLabs = allBasicInfo_filt_sp_mat_selected --> <!-- rownames(allBasicInfo_filt_sp_mat_noLabs) = NULL --> <!-- colnames(allBasicInfo_filt_sp_mat_noLabs) = NULL --> <!-- rowAnnoDf = metaReSelected[,c("hrpCall", "hrp3DeletionPattern", "isolate", "country", "region", "secondaryRegion")] %>% rename(continent = secondaryRegion) %>% as.data.frame() --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr08 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_08_", colnames(allBasicInfo_filt_sp_mat))] --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr11 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_11_", colnames(allBasicInfo_filt_sp_mat))] --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr13 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_13_", colnames(allBasicInfo_filt_sp_mat))] --> <!-- # topAnnoDf = regions[,c("inGene", "genomicRegion", "chrom")] %>% as.data.frame() --> <!-- topAnnoDf = regions[,c("inGene", "genomicRegion", "chrom")] %>% as.data.frame() --> <!-- topAnnoDf_chr08 = topAnnoDf %>% filter(grepl("_08_",chrom)) %>% select(-chrom) --> <!-- topAnnoDf_chr11 = topAnnoDf %>% filter(grepl("_11_",chrom)) %>% select(-chrom) --> <!-- topAnnoDf_chr13 = topAnnoDf %>% filter(grepl("_13_",chrom)) %>% select(-chrom) --> <!-- annotationTextSize = 20 --> <!-- topAnno_chr08 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr08, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left" --> <!-- ) --> <!-- topAnno_chr11 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr11, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- show_annotation_name = F --> <!-- ) --> <!-- topAnno_chr13 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr13, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- show_annotation_name = F --> <!-- ) --> <!-- sideAnno = rowAnnotation( --> <!-- df = rowAnnoDf, --> <!-- col = rowAnnoColors, --> <!-- gp = gpar(col = "grey10"), --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ) --> <!-- ) --> <!-- library(dendsort) --> <!-- clustering = --> <!-- # row_dend = dendsort(hclust(dist(allBasicInfo_filt_sp_mat_noLabs, method = "euclidean"), method = "complete")) --> <!-- # row_dend$method = "euclidean"; row_dend$dist.method = "complete" --> <!-- # col_dend = dendsort(hclust(dist(t(allBasicInfo_filt_sp_mat_noLabs), method = "euclidean"), method = "complete")) --> <!-- # col_dend$method = "euclidean"; col_dend$dist.method = "complete" --> <!-- row_dend = dendsort(hclust(dist(allBasicInfo_filt_sp_mat_noLabs))) --> <!-- allCoverageHeatMap_chr08 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr08, --> <!-- cluster_columns = F, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr08, --> <!-- left_annotation = sideAnno, --> <!-- cluster_rows = row_dend, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr08", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- allCoverageHeatMap_chr11 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr11, --> <!-- cluster_columns = F, --> <!-- cluster_rows = row_dend, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr11, --> <!-- # left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr11", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- allCoverageHeatMap_chr13 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr13, --> <!-- cluster_columns = F, --> <!-- cluster_rows = row_dend, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr13, --> <!-- # left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr13", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- hms_list = allCoverageHeatMap_chr08 + allCoverageHeatMap_chr11 + allCoverageHeatMap_chr13 --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen --> <!-- #| fig-width: 30 --> <!-- #| fig-height: 20 --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- ``` --> <!-- ```{r} --> <!-- pdf("allHeatmap_byChrom_hrp3_deleted_filtered.pdf", useDingbats = F, width = 30, height = 20) --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- dev.off() --> <!-- ``` --> <!-- ### Hrp2- and Hrp3- --> <!-- ```{r} --> <!-- allBasicInfo_filt_sp_mat_selected = allBasicInfo_filt_sp_mat[rownames(allBasicInfo_filt_sp_mat) %in% metaSelected_hrp2_and_hrp3_deleted$sample, ] --> <!-- metaReSelected = meta[match(rownames(allBasicInfo_filt_sp_mat_selected), meta$sample), ] --> <!-- library(circlize) --> <!-- col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3))) --> <!-- allBasicInfo_filt_sp_mat_noLabs = allBasicInfo_filt_sp_mat_selected --> <!-- rownames(allBasicInfo_filt_sp_mat_noLabs) = NULL --> <!-- colnames(allBasicInfo_filt_sp_mat_noLabs) = NULL --> <!-- rowAnnoDf = metaReSelected[,c("hrpCall", "hrp3DeletionPattern", "isolate", "country", "region", "secondaryRegion")] %>% rename(continent = secondaryRegion) %>% as.data.frame() --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr08 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_08_", colnames(allBasicInfo_filt_sp_mat))] --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr11 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_11_", colnames(allBasicInfo_filt_sp_mat))] --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr13 = allBasicInfo_filt_sp_mat_noLabs[,grepl("_13_", colnames(allBasicInfo_filt_sp_mat))] --> <!-- # topAnnoDf = regions[,c("inGene", "genomicRegion", "chrom")] %>% as.data.frame() --> <!-- topAnnoDf = regions[,c("inGene", "genomicRegion", "chrom")] %>% as.data.frame() --> <!-- topAnnoDf_chr08 = topAnnoDf %>% filter(grepl("_08_",chrom)) %>% select(-chrom) --> <!-- topAnnoDf_chr11 = topAnnoDf %>% filter(grepl("_11_",chrom)) %>% select(-chrom) --> <!-- topAnnoDf_chr13 = topAnnoDf %>% filter(grepl("_13_",chrom)) %>% select(-chrom) --> <!-- annotationTextSize = 20 --> <!-- topAnno_chr08 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr08, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left" --> <!-- ) --> <!-- topAnno_chr11 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr11, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- show_annotation_name = F --> <!-- ) --> <!-- topAnno_chr13 = HeatmapAnnotation( --> <!-- df = topAnnoDf_chr13, --> <!-- col = topAnnoColors, --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- annotation_name_side = "left", --> <!-- show_annotation_name = F --> <!-- ) --> <!-- sideAnno = rowAnnotation( --> <!-- df = rowAnnoDf, --> <!-- col = rowAnnoColors, --> <!-- gp = gpar(col = "grey10"), --> <!-- annotation_name_gp = gpar(fontsize = annotationTextSize), --> <!-- annotation_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ) --> <!-- ) --> <!-- library(dendsort) --> <!-- clustering = --> <!-- # row_dend = dendsort(hclust(dist(allBasicInfo_filt_sp_mat_noLabs, method = "euclidean"), method = "complete")) --> <!-- # row_dend$method = "euclidean"; row_dend$dist.method = "complete" --> <!-- # col_dend = dendsort(hclust(dist(t(allBasicInfo_filt_sp_mat_noLabs), method = "euclidean"), method = "complete")) --> <!-- # col_dend$method = "euclidean"; col_dend$dist.method = "complete" --> <!-- row_dend = dendsort(hclust(dist(allBasicInfo_filt_sp_mat_noLabs))) --> <!-- allCoverageHeatMap_chr08 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr08, --> <!-- cluster_columns = F, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr08, --> <!-- left_annotation = sideAnno, --> <!-- cluster_rows = row_dend, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr08", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- allCoverageHeatMap_chr11 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr11, --> <!-- cluster_columns = F, --> <!-- cluster_rows = row_dend, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr11, --> <!-- # left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr11", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- allCoverageHeatMap_chr13 = Heatmap( --> <!-- allBasicInfo_filt_sp_mat_noLabs_chr13, --> <!-- cluster_columns = F, --> <!-- cluster_rows = row_dend, --> <!-- col = col_fun, --> <!-- name = "coverage", --> <!-- top_annotation = topAnno_chr13, --> <!-- # left_annotation = sideAnno, --> <!-- row_dend_width = unit(5, "cm"), --> <!-- column_dend_height = unit(5, "cm"), --> <!-- heatmap_legend_param = list( --> <!-- labels_gp = gpar(fontsize = annotationTextSize), --> <!-- title_gp = gpar(fontsize = annotationTextSize, fontface = "bold") --> <!-- ), --> <!-- column_title = "Chr13", --> <!-- column_title_gp = gpar(fontsize = 30, fontface = "bold") --> <!-- ) --> <!-- hms_list = allCoverageHeatMap_chr08 + allCoverageHeatMap_chr11 + allCoverageHeatMap_chr13 --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen --> <!-- #| fig-width: 30 --> <!-- #| fig-height: 20 --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- ``` --> <!-- ```{r} --> <!-- pdf("allHeatmap_byChrom_hrp2_and_hrp3_deleted_filtered.pdf", useDingbats = F, width = 30, height = 20) --> <!-- draw(hms_list, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "left", annotation_legend_side = "left", ht_gap = unit(2, "cm")) --> <!-- dev.off() --> <!-- ``` -->