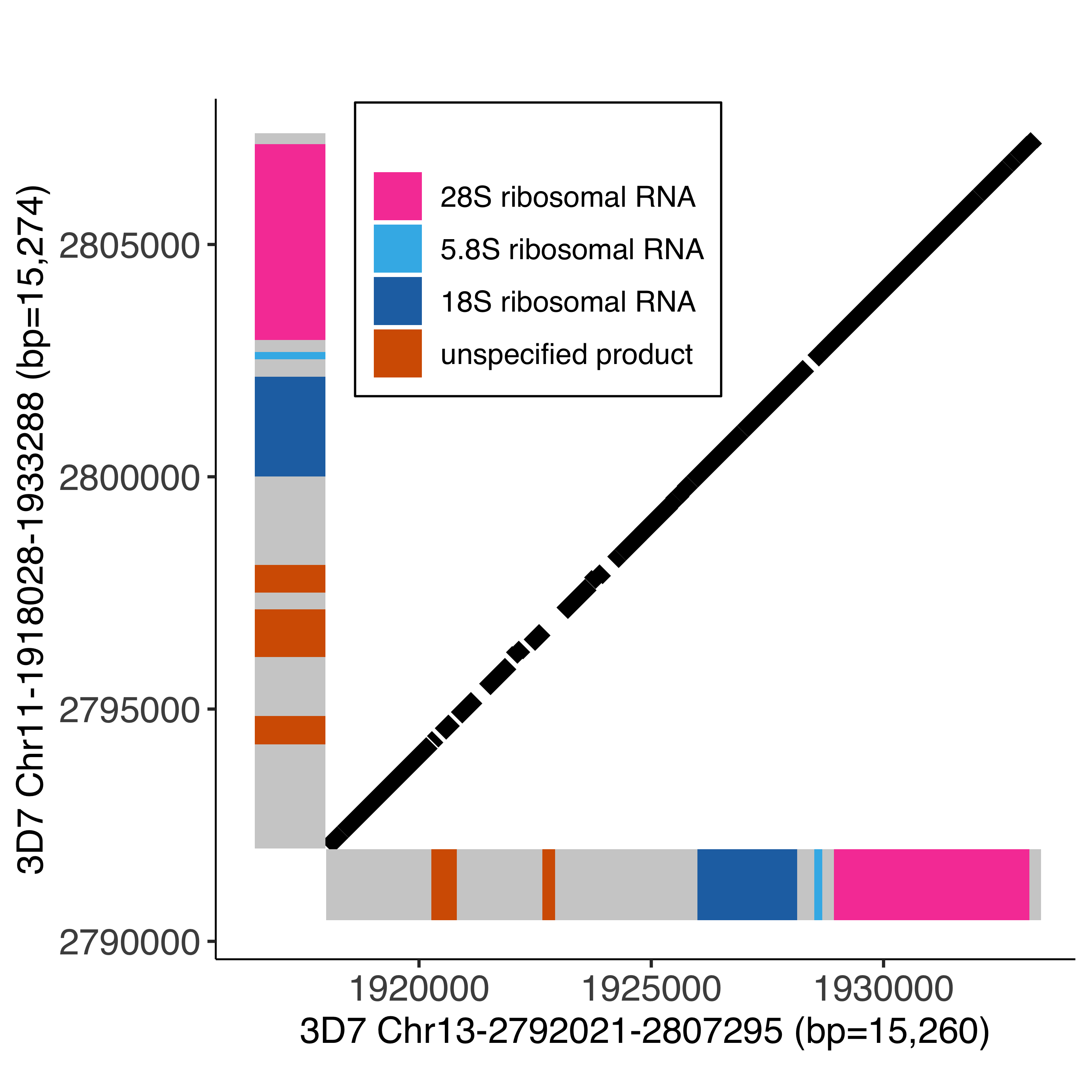
--- title: Finding unqiue matches interchromosmal --- ```{r setup, echo=FALSE, message=FALSE} source("../common.R") ``` # Comparing unique stretches between genomes ```{bash, eval = F} elucidator gffToBedByFeature --gff /tank/data/plasmodium/genomes/Pf3D7_versions/2015-06-18/info/gff/Pf3D7.gff --features gene,psuedogene,protein_coding_gene,ncRNA_gene --out Pf3D7_genes.bed --overWrite elucidator splitColumnContainingMeta --file Pf3D7_genes.bed --column col.6 --delim tab --removeEmptyColumn --overWrite --out Pf3D7_genes.tab.txt --addHeader ``` ```{r} = readr:: read_tsv ("surroundingRegionsMaterials/chromLengths/Pf3D7.txt" , col_names = c ("chrom" , "length" ))= readr:: read_tsv ("nucmerResults/genes/Pf3D7_genes.tab.txt" ) %>% mutate (description = gsub (" \\ +" , " " , description))``` # nucmer ## Running nucmer ```{bash, eval = F} for CHR in 01 02 03 04 05 06 07 08 09 10 11 12 13 14; do elucidator extractByName --fasta /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta --trimAtWhiteSpace --names Pf3D7_${CHR}_v3 --out Pf3D7_${CHR}_v3.fasta --overWrite; done; ``` ```{r} = c ()for (chr1 in seq (1 , 14 , 1 )){for (chr2 in seq (chr1, 14 , 1 )){if (chr1 != chr2){= stringr:: str_pad (chr1, width = 2 , pad = "0" )= stringr:: str_pad (chr2, width = 2 , pad = "0" )= paste0 ("mummer -mum -b -c -F -l 31 Pf3D7_" , chr1Name, "_v3.fasta Pf3D7_" , chr2Name, "_v3.fasta 2> mumer_Pf3D7_" , chr1Name, "_v3_vs_Pf3D7_" , chr2Name, "_v3.log | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_exactUniqueMatches_minlen31.tsv && nucmer -mum -b 100 -l 31 Pf3D7_" , chr1Name, "_v3.fasta Pf3D7_" , chr2Name, "_v3.fasta --prefix Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_nucmer 2> nucmer_Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_v3.log && show-coords -T -l -c -H Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_nucmer.delta | sort | uniq | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_nucmer.delta.bed && elucidator splitColumnContainingMeta --file Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --replacementHeader \" #chrom,start,end,name,length,strand \" --out Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_nucmer.delta.tsv" )= c (allNucmerCmds,else { = stringr:: str_pad (chr1, width = 2 , pad = "0" )= stringr:: str_pad (chr2, width = 2 , pad = "0" )= paste0 ("mummer -mum -r -c -F -l 31 Pf3D7_" , chr1Name, "_v3.fasta Pf3D7_" , chr2Name, "_v3.fasta 2> mumer_Pf3D7_" , chr1Name, "_v3_vs_Pf3D7_" , chr2Name, "_v3.log | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_exactUniqueMatches_minlen31.tsv && nucmer --reverse -mum -b 100 -l 31 Pf3D7_" , chr1Name, "_v3.fasta Pf3D7_" , chr2Name, "_v3.fasta --prefix Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_nucmer 2> nucmer_Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_v3.log && show-coords -T -l -c -H Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_nucmer.delta | sort | uniq | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_nucmer.delta.bed && elucidator splitColumnContainingMeta --file Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --replacementHeader \" #chrom,start,end,name,length,strand \" --out Pf3D7_" , chr1Name, "_vs_Pf3D7_" , chr2Name, "_nucmer.delta.tsv" )= c (allNucmerCmds, cat (allNucmerCmds, sep = " \n " , file = "allNucmerCmds.txt" )``` ```{bash, eval = F} nohup elucidator runMultipleCommands --cmdFile allNucmerCmds.txt --numThreads 10 --raw & elucidator rBind --contains _nucmer.delta.tsv --delim tab --header --overWrite --out allNucmerResults.tsv elucidator rBind --contains _exactUniqueMatches_minlen31.tsv --delim tab --header --overWrite --out allMummerResults.tsv ``` ## Processing results ### nucmer ```{r} = readr:: read_tsv ("nucmerResults/allNucmerResults.tsv" )= allNucmerResults %>% mutate (genomicID = paste0 (` #chrom ` , "-" , start, "-" , end, "-" , ifelse (strand == "+" , "for" , "rev" ))) %>% mutate (queryGenomicID = paste0 (queryName, "-" , actualStart, "-" , actualEnd, "-for" )) %>% group_by (genomicID, queryGenomicID) %>% mutate (wholeID = paste0 (sort (c (genomicID, queryGenomicID)), collapse = "__" )) %>% group_by (wholeID) %>% mutate (wholeID_n = n ()) %>% arrange (desc (length)) %>% mutate (queryStrand = "+" )= allNucmerResults %>% ungroup () %>% select (` #chrom ` , start, end, wholeID, length, strand)= allNucmerResults %>% ungroup () %>% mutate (queryLength = actualEnd - actualStart) %>% select (queryName, actualStart, actualEnd, wholeID, queryLength, queryStrand)colnames (allNucmerResults_ref_out) = c ("#chrom" , "start" , "end" , "name" , "length" , "strand" )colnames (allNucmerResults_query_out) = c ("#chrom" , "start" , "end" , "name" , "length" , "strand" )= bind_rows (%>% arrange (desc (length), name)= allNucmerResults_combined_out %>% filter (length > 1000 )write_tsv (allNucmerResults_combined_out_1k, "nucmerResults/allNucmerResults1k.bed" )``` ```{bash, eval = F} elucidator bedGetIntersectingGenesInGff --gff /tank/data/plasmodium/genomes/Pf3D7_versions/2015-06-18/info/gff/Pf3D7.gff --extraAttributes description --overWrite --bed allNucmerResults1k.bed --out allNucmerResults1k_withGeneInfo.bed --selectFeatures gene,psuedogene,protein_coding_gene,ncRNA_gene elucidator getOverlappingBedRegions --bed genes/Pf3D7_genes.bed --intersectWithBed allNucmerResults1k.bed | cut -f1-7 > genes/Pf3D7_genes_in_allNucmerResults1k.bed elucidator splitColumnContainingMeta --file genes/Pf3D7_genes_in_allNucmerResults1k.bed --delim tab --removeEmptyColumn --addHeader --column col.6 --overWrite --out genes/Pf3D7_genes_in_allNucmerResults1k.tab.txt ``` ```{r} = readr:: read_tsv ("nucmerResults/allNucmerResults1k_withGeneInfo.bed" , col_names = F)``` ```{r} #| results: asis #| echo: false cat (createDownloadLink ("nucmerResults/allNucmerResults1k_withGeneInfo.bed" ))``` ### mummer ```{r} = readr:: read_tsv ("nucmerResults/allMummerResults.tsv" ) %>% arrange (desc (length)) %>% mutate (genomicID = paste0 (` #target ` , "-" , targetStart, "-" , targetEnd, "-" , ifelse (strand == "+" , "for" , "rev" ))) %>% mutate (queryGenomicID = paste0 (query, "-" , queryStart, "-" , queryEnd, "-for" )) %>% group_by (genomicID, queryGenomicID) %>% mutate (wholeID = paste0 (sort (c (genomicID, queryGenomicID)), collapse = "__" ))= allMummerResults %>% ungroup () %>% select (` #target ` , targetStart, targetEnd, wholeID, length, strand)= allMummerResults %>% ungroup () %>% select (query, queryStart, queryEnd, wholeID, length) %>% mutate (strand = "+" )colnames (allMummerResults_target) = c ("#chrom" , "start" , "end" , "name" , "length" , "strand" )colnames (allMummerResults_query) = c ("#chrom" , "start" , "end" , "name" , "length" , "strand" )= bind_rows (= allMummerResults_combined %>% filter (length >= 50 )write_tsv (allMummerResults_combined_filtMinLen50, "nucmerResults/allMummerResultsExpandedFiltMinLen50.bed" )``` ```{bash, eval = F} elucidator bedGetIntersectingGenesInGff --gff /tank/data/plasmodium/genomes/Pf3D7_versions/2015-06-18/info/gff/Pf3D7.gff --extraAttributes description --overWrite --bed allMummerResultsExpandedFiltMinLen50.bed --out allMummerResultsExpandedFiltMinLen50_withGeneInfo.bed --selectFeatures gene,psuedogene,protein_coding_gene,ncRNA_gene ``` ```{r} #| results: asis #| echo: false cat (createDownloadLink ("nucmerResults/allMummerResultsExpandedFiltMinLen50_withGeneInfo.bed" ))``` ```{r} = readr:: read_tsv ("nucmerResults/allMummerResultsExpandedFiltMinLen50_withGeneInfo.bed" , col_names = F)``` ```{bash, eval = F} elucidator getOverlappingBedRegions --bed allMummerResultsExpandedFiltMinLen50.bed --intersectWithBed allNucmerResults1k.bed --overWrite --out allMummerResultsExpandedFiltMinLen50_intersectedWithNucmer.bed ``` ```{r} #| fig-column: screen #| column: screen-inset-shaded = readr:: read_tsv ("nucmerResults/allMummerResultsExpandedFiltMinLen50_intersectedWithNucmer.bed" , col_names = F, skip = 1 )colnames (allMummerResults_intersectedWithNucmer) = c ("chrom" , "start" , "end" , "name" , "length" , "strand" , "group" )= allMummerResults_intersectedWithNucmer %>% mutate (group = strsplit (group, split = "," )) %>% unnest (group)= allMummerResults_intersectedWithNucmer %>% # filter(name %in% c("Pf3D7_05_v3-0-23704-for__Pf3D7_13_v3-435-24139-for", # "Pf3D7_13_v3-1431002-1439191-rev__Pf3D7_13_v3-1440439-1448628-for", # "Pf3D7_04_v3-265-5013-rev__Pf3D7_06_v3-1411966-1416714-for")) %>% separate (group, into = c ("chrom1_section" , "chrom2_section" ), remove = F, sep = "__" ) %>% separate (chrom1_section,into = c ("chrom1_section_chrom" , "chrom1_section_start" , "chrom1_section_end" , "chrom1_section_strand" ),remove = F, sep = "-" , convert = T) %>% mutate (chrom1_section_strand = ifelse (chrom1_section_strand == "for" , "+" , "-" )) %>% separate (chrom2_section,into = c ("chrom2_section_chrom" , "chrom2_section_start" , "chrom2_section_end" , "chrom2_section_strand" ),remove = F, sep = "-" , convert = T) %>% mutate (chrom2_section_strand = ifelse (chrom2_section_strand == "for" , "+" , "-" )) %>% mutate (chrom2_section_len = chrom2_section_end - chrom2_section_start, chrom1_section_len = chrom1_section_end - chrom1_section_start) %>% mutate (other = gsub (".*__" , "" , name)) %>% separate (other, remove = F, sep = "-" , convert = T, into = c ("other_chrom" , "other_start" , "other_end" , "other_strand" )) %>% mutate (other_strand = ifelse (other_strand == "for" , "+" , "-" )) %>% filter (strand == chrom1_section_strand, == chrom2_section_strand, == chrom1_section_chrom, == chrom2_section_chrom, >= chrom1_section_start, <= chrom1_section_end, >= chrom2_section_start, <= chrom2_section_end) %>% group_by (name, group) %>% mutate (allBases = list (seq (start, end)))= allMummerResults_intersectedWithNucmer_mod%>% group_by (group) %>% mutate (totalBases = length (unique (unlist (allBases)))) %>% arrange (desc (totalBases)) %>% mutate (chrom1_fracConserved = totalBases/ chrom1_section_len, chrom2_fracConserved = totalBases/ chrom2_section_len) %>% ungroup ()= allMummerResults_intersectedWithNucmer_mod_withSum %>% select (group, totalBases, starts_with ("chrom1_" ), starts_with ("chrom2_" )) %>% unique ()create_dt (allMummerResults_intersectedWithNucmer_mod_withSum_sel)``` ## Visualizing the top 10 results ```{r} = list ()for (grouping in unique (allMummerResults_intersectedWithNucmer_mod_withSum$ group)[1 : 10 ]){= allMummerResults_intersectedWithNucmer_mod_withSum %>% filter (grouping == group)= pf3d7Genes %>% filter (col.0 == conservedInfo_current$ chrom[1 ], .1 >= conservedInfo_current$ chrom1_section_start[1 ] | col.2 >= conservedInfo_current$ chrom1_section_start[1 ], .1 <= conservedInfo_current$ chrom1_section_end[1 ] | col.2 <= conservedInfo_current$ chrom1_section_end[1 ])= pf3d7Genes %>% filter (col.0 == conservedInfo_current$ other_chrom[1 ], .1 >= conservedInfo_current$ chrom2_section_start[1 ] | col.2 >= conservedInfo_current$ chrom2_section_start[1 ], .1 <= conservedInfo_current$ chrom2_section_end[1 ] | col.2 <= conservedInfo_current$ chrom2_section_end[1 ])= pf3d7Chroms %>% filter (chrom %in% unique (c (conservedInfo_current$ chrom, conservedInfo_current$ other_chrom)))= pf3d7Chroms_filt %>% mutate (chrom = factor (chrom, levels = c (.$ chrom)))= conservedInfo_current %>% group_by (chrom1_section_chrom, chrom1_section_start, chrom1_section_end, %>% summarise (total = n (), revCompSum = sum (strand == "-" )) %>% mutate (chrom1RevCompFrac = revCompSum/ total) %>% mutate (chrom1RevComp = ifelse (chrom1RevCompFrac > 0.5 , T, F))= conservedInfo_current_sum %>% mutate (chrom1_section_chrom = factor (chrom1_section_chrom, levels = c (pf3d7Chroms_filt$ chrom))) %>% mutate (chrom2_section_chrom = factor (chrom2_section_chrom, levels = c (pf3d7Chroms_filt$ chrom)))#cat(c(grouping, unique(conservedInfo_current$chrom1_section_len)), sep = "\n") = ggplot (conservedInfo_current) + geom_segment (aes (x = start,xend = end,y = ifelse (strand == "-" , other_end, other_start),yend = ifelse (strand == "-" , other_start, other_end),color = strand+ geom_rect (data = pf3d7Genes_chrom1, aes (xmin = col.1 , xmax = col.2 , ymax = conservedInfo_current$ chrom2_section_start[1 ] - conservedInfo_current$ chrom2_section_len[1 ] * 0.001 , ymin = conservedInfo_current$ chrom2_section_start[1 ] - conservedInfo_current$ chrom2_section_len[1 ] * 0.01 , fill = description, ID = ID)) + geom_rect (data = pf3d7Genes_chrom2, aes (ymin = col.1 , ymax = col.2 , xmax = conservedInfo_current$ chrom1_section_start[1 ] - conservedInfo_current$ chrom1_section_len[1 ] * 0.001 , xmin = conservedInfo_current$ chrom1_section_start[1 ] - conservedInfo_current$ chrom1_section_len[1 ] * 0.01 , fill = description, ID = ID )) + labs (title = grouping, y = pf3d7Genes_chrom2$ col.0 [1 ], x = pf3d7Genes_chrom1$ col.0 [1 ]) + sofonias_theme + coord_equal () + scale_color_manual (values = c ("-" = "#AA0A3C" , "+" = "#0AB45A" ));if (conservedInfo_current_sum$ chrom1RevComp[1 ]){= currentPlot + scale_y_reverse ()= ggplotly (currentPlot)paste0 (grouping, "-fullView" )]] = ggplotly (ggplot () + geom_rect (data = pf3d7Chroms_filt,aes (xmin = 0 ,xmax = length,ymin = as.numeric (chrom) - 0.4 ,ymax = as.numeric (chrom) + 0.4 fill = "grey75" + scale_y_continuous (breaks = 1 : (nrow (pf3d7Chroms_filt)),labels = levels (pf3d7Chroms_filt$ chrom)+ + geom_rect (data = conservedInfo_current_sum,aes (xmin = chrom1_section_start,xmax = chrom1_section_end,ymin = as.numeric (chrom1_section_chrom) - 0.4 ,ymax = as.numeric (chrom1_section_chrom) + 0.4 ,fill = chrom1RevComp+ geom_rect (data = conservedInfo_current_sum,aes (xmin = chrom2_section_start,xmax = chrom2_section_end,ymin = as.numeric (chrom2_section_chrom) - 0.4 ,ymax = as.numeric (chrom2_section_chrom) + 0.4 fill = "#0AB45A" + scale_fill_manual (values = c ("TRUE" = "#AA0A3C" , "FALSE" = "#0AB45A" ))``` ```{r} #| fig-column: screen-inset #| column: screen-inset #| results: asis #| fig-width: 15 #| fig-height: 25 cat (create_tabsetOfHtmlWidgets (listOfPlots))#htmltools::tagList(listOfPlots) ``` ```{r} #| fig-column: body-outset #| column: body-outset = allMummerResults_intersectedWithNucmer_mod_withSum %>% group_by (chrom1_section_chrom, chrom2_section_chrom) %>% summarise (total = sum (totalBases)) %>% arrange (desc (total))create_dt (allMummerResults_intersectedWithNucmer_mod_withSum_sum)``` # View All Regions ```{r, fig.width=20} pf3d7Chroms = pf3d7Chroms %>% mutate(chrom = factor(chrom, levels = c(.$chrom))) allNucmerResults1k_withGeneInfo = allNucmerResults1k_withGeneInfo %>% mutate(X1 = factor(X1, levels = c(pf3d7Chroms$chrom))) %>% rename(len = X5, name = X4) pf3d7Genes = readr::read_tsv("nucmerResults/genes/Pf3D7_genes_in_allNucmerResults1k.tab.txt") %>% mutate(rawGeneDescription = description) %>% mutate(description = gsub("\\+", " ", description))%>% mutate(gene = ifelse(grepl("PF3D7_0831800", description), "HRP II", "other")) %>% mutate(gene = ifelse(grepl("PF3D7_1372200", description), "HRP III", gene))%>% mutate(description = gsub("unknownfunction", "unknown function", description))%>% mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>% mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>% mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>% mutate(description = ifelse("membrane associated erythrocyte binding-like protein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>% mutate(description = ifelse("membrane associated erythrocyte binding-likeprotein"==description, "merozoite adhesive erythrocytic binding protein", description)) %>% mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description)) %>% mutate(description = gsub(",putative", ", putative", description)) %>% mutate(description = gsub(",pseudogene", ", pseudogene", description))%>% mutate(description = gsub("unknownfunction", "unknown function", description))%>% mutate(description = gsub("conserved protein, unknown function", "conserved Plasmodium protein, unknown function", description)) %>% mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>% mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>% mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>% mutate(description = ifelse("membrane associated histidine-rich protein 1"==description, "membrane associated histidine-rich protein", description))%>% mutate(description = gsub(",pseudogene", ", pseudogene", description))%>% mutate(description = gsub("unknownfunction", "unknown function", description))%>% mutate(description = gsub(" \\(SURFIN", "\\(SURFIN", description)) %>% mutate(description = ifelse(grepl("Plasmodium exported protein", description), "Plasmodium exported protein (PHIST)", description)) %>% mutate(description = ifelse("erythrocyte membrane protein 1 (PfEMP1), exon 2"==description, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene", description)) %>% mutate(description = ifelse(grepl("sporozoite and liver stage tryptophan-rich protein, putative", description), "tryptophan/threonine-rich antigen", description))%>% mutate(description = ifelse(grepl("CRA domain-containing protein, putative", description), "conserved Plasmodium protein, unknown function", description))%>% mutate(description = gsub(",pseudogene", ", pseudogene", description)) %>% mutate(description = gsub("surfaceantigen", "surface antigen", description)) %>% mutate(description = gsub("Tetratricopeptide repeat, putative", "tetratricopeptide repeat protein, putative", description)) %>% mutate(description = gsub("transmembraneprotein", "transmembrane protein", description)) %>% mutate(description = ifelse(grepl("PfEMP1", description) & grepl("pseudogene", description), "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>% mutate(description = gsub("PIR protein", "stevor", description)) %>% mutate(description = gsub("erythrocyte membrane protein 1-like", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description)) %>% mutate(description = gsub("acidic terminal segments, variant surface antigen of PfEMP1, putative", "erythrocyte membrane protein 1 (PfEMP1), pseudogene", description))%>% mutate(description = ifelse(grepl("CoA binding protein", description, ignore.case = T), "acyl-CoA binding protein", description)) %>% mutate(description = ifelse(grepl("transfer RNA", description) | grepl("tRNA", description), "tRNA", description))%>% mutate(description = ifelse(grepl("cytoadherence", description), "CLAG", description))%>% mutate(description = ifelse(grepl("surface-associated interspersed protein", description), "SURFIN", description))%>% mutate(description = ifelse(grepl("SURFIN", description), "SURFIN", description)) %>% mutate(description = ifelse(grepl("stevor-like", description), "stevor, pseudogene", description)) %>% mutate(description = ifelse(grepl("non-coding RNA", description), "unspecified product", description)) pf3d7Genes = pf3d7Genes %>% rename(chrom = col.0, start = col.1, end = col.2, name = col.3, len = col.4, strand = col.5) pf3d7Genes = pf3d7Genes %>% mutate(chrom = factor(chrom, levels = c(pf3d7Chroms$chrom))) descriptColors = scheme$hex(length(sort(unique(c(pf3d7Genes$description))))) names(descriptColors) = sort(unique(c(pf3d7Genes$description))) ``` ```{r} #| fig-column: screen #| column: screen-inset-shaded #| fig-height: 15 #| fig-width: 20 ggplotly (ggplot () + geom_rect (data = pf3d7Chroms,aes (xmin = 0 ,xmax = length,ymin = as.numeric (chrom) - 0.4 ,ymax = as.numeric (chrom) + 0.4 fill = "grey75" + scale_y_continuous (breaks = 1 : (nrow (pf3d7Chroms)),labels = levels (pf3d7Chroms$ chrom)+ + geom_rect (data = allNucmerResults1k_withGeneInfo,aes (xmin = X2,xmax = X3,ymin = as.numeric (X1) - 0.4 ,ymax = as.numeric (X1) + 0.4 ,fill = X6, len = len, name = name+ geom_rect (data = pf3d7Genes,aes (xmin = start,xmax = end,ymin = as.numeric (chrom) - 0.5 ,ymax = as.numeric (chrom) - 0.4 ,fill = description, ID = ID, feature = feature, chrom = chrom, start = start, end = end, strand = strand,name = name+ scale_fill_manual (values = c (c ("-" = "#AA0A3C50" , "+" = "#3DB7E950" ), ``` ## chr11 chr13 rRNA - Pf3D7_11_v3-1918005-1933391-for__Pf3D7_13_v3-2791996-2807398-for ```{r} #| fig-column: screen = "Pf3D7_11_v3-1918005-1933391-for__Pf3D7_13_v3-2791996-2807398-for" = allMummerResults_intersectedWithNucmer_mod_withSum %>% filter (grouping == group) %>% filter (length >= 100 )= conservedInfo_current %>% group_by (chrom) %>% summarise (start = min (start), end = max (end)) %>% mutate (name = paste0 (chrom, "-" , start, "-" , end), len = end - start, strand = "+" )= conservedInfo_current %>% group_by (other_chrom) %>% summarise (start = min (other_start), end = max (other_end)) %>% rename (chrom = other_chrom) %>% mutate (name = paste0 (chrom, "-" , start, "-" , end), len = end - start, strand = "+" )# Re-cut region to the stretches of unqiue sequences without the nucmer expansion = conservedInfo_current_chrom_sum %>% bind_rows (conservedInfo_current_other_sum)= pf3d7Genes %>% filter (chrom == conservedInfo_current$ chrom[1 ], >= conservedInfo_current$ chrom1_section_start[1 ], <= conservedInfo_current$ chrom1_section_end[1 ]) %>% filter (start < 1933277 )= pf3d7Genes %>% filter (chrom == conservedInfo_current$ other_chrom[1 ], >= conservedInfo_current$ chrom2_section_start[1 ], <= conservedInfo_current$ chrom2_section_end[1 ])write_tsv (conservedInfo_current, "conservedInfo_between_11_and_13_sharedRegion_nucmer.tsv" )= ggplot (conservedInfo_current) + geom_segment (aes (x = start,xend = end,y = other_start,yend = other_endlinewidth = 2.5 ,color = "black" + geom_rect (ymin = conservedInfo_current$ chrom2_section_start[1 ],ymax = conservedInfo_current$ chrom2_section_start[1 ] + conservedInfo_current$ chrom2_section_len[1 ], xmin = conservedInfo_current$ chrom1_section_start[1 ] - conservedInfo_current$ chrom1_section_len[1 ] * 0.1 ,xmax = conservedInfo_current$ chrom1_section_start[1 ] - conservedInfo_current$ chrom1_section_len[1 ] * 0.001 , fill = "grey81" ) + geom_rect (ymax = conservedInfo_current$ chrom2_section_start[1 ] - conservedInfo_current$ chrom2_section_len[1 ] * 0.001 , ymin = conservedInfo_current$ chrom2_section_start[1 ] - conservedInfo_current$ chrom2_section_len[1 ] * 0.1 , xmin = conservedInfo_current$ chrom1_section_start[1 ],xmax = conservedInfo_current$ chrom1_section_start[1 ] + conservedInfo_current$ chrom1_section_len[1 ], fill = "grey81" ) + geom_rect (data = pf3d7Genes_chrom1, aes (xmin = start, xmax = end, ymax = conservedInfo_current$ chrom2_section_start[1 ] - conservedInfo_current$ chrom2_section_len[1 ] * 0.001 , ymin = conservedInfo_current$ chrom2_section_start[1 ] - conservedInfo_current$ chrom2_section_len[1 ] * 0.1 , fill = description)) + geom_rect (data = pf3d7Genes_chrom2, aes (ymin = start, ymax = end, xmax = conservedInfo_current$ chrom1_section_start[1 ] - conservedInfo_current$ chrom1_section_len[1 ] * 0.001 , xmin = conservedInfo_current$ chrom1_section_start[1 ] - conservedInfo_current$ chrom1_section_len[1 ] * 0.1 , fill = description)) + labs (title = "" , x = "3D7 Chr13-2792021-2807295 (bp=15,260)" , y = "3D7 Chr11-1918028-1933288 (bp=15,274)" , fill = "" ) + + coord_equal () + # scale_fill_tableau() + scale_fill_manual (values = c ( "#F748A5" , "#3DB7E9" , "#2271B2" , "#D55E00" ), breaks = c ("28S ribosomal RNA" , "5.8S ribosomal RNA" , "18S ribosomal RNA" , "unspecified product" )) + guides (fill= guide_legend (ncol = 1 ,byrow= TRUE )) + theme (legend.position = c (0.375 , 0.825 ), panel.border = element_blank (), legend.background = element_blank (),legend.box.background = element_rect (colour = "black" ))``` ```{r} #| fig-column: screen #| column: screen-inset-shaded #| fig-width: 5 #| fig-height: 5 print (conservedInfo_current_plot)``` ```{r} cairo_pdf ("nucmer_shared_3D7chr11-3D7chr13_region.pdf" , width = 5 , height = 5 )print (conservedInfo_current_plot)dev.off ()``` ```{r} #| results: asis #| echo: false cat (createDownloadLink ("shared_3D7chr11-3D7chr13_region.pdf" ))``` ### Comparison of region ```{bash, eval = F} elucidator bedRenameWithCoords --bed ../../../../sharedBetween11_and_13/investigatingChrom11Chrom13/shared_11_13_region.bed --out renamed_shared_11_13_region.bed elucidator getFastaWithBed --overWrite --twoBit /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.2bit --bed renamed_shared_11_13_region.bed --out renamed_shared_11_13_region.fasta elucidator compareToRef --fasta renamed_shared_11_13_region.fasta --out renamed_shared_11_13_region_comparison --ref renamed_shared_11_13_region.fasta elucidator trimToLen --length 7981 --fasta renamed_shared_11_13_region.fasta --overWrite --out trimmed_to_7981.fasta elucidator compareToRef --fasta trimmed_to_7981.fasta --out trimmed_to_7981_comparison --ref trimmed_to_7981.fasta elucidator trimFront --forwardBases 7891 --fasta renamed_shared_11_13_region.fasta --overWrite --out trimmed_from_7981.fasta elucidator compareToRef --fasta trimmed_from_7981.fasta --out trimmed_from_7981_comparison --ref trimmed_from_7981.fasta ``` ```{r} = bind_rows (:: read_tsv ("surroundingRegionsMaterials/interchromosomalComparison/renamed_shared_11_13_region_comparison.txt" ,%>% mutate (region = "wholeDupRegion" ),:: read_tsv ("surroundingRegionsMaterials/interchromosomalComparison/trimmed_to_7981_comparison.txt" ,%>% mutate (region = "prior_to_rRNA_region" ),:: read_tsv ("surroundingRegionsMaterials/interchromosomalComparison/trimmed_from_7981_comparison.txt" ,%>% mutate (region = "rRNA_region" )create_dt (comps)``` # All rRNA regions ```{r} = readr:: read_tsv ("nucmerResults/genes/Pf3D7_genes.tab.txt" ) %>% mutate (description = gsub (" \\ +" , " " , description))= readr:: read_tsv ("nucmerResults/allNucmerResults1k_withGeneInfo.bed" , col_names = F)= nucmerResults_withGeneInfo %>% filter (grepl ("ribosomal.*RNA" , X7)) %>% arrange (desc (X5))= list ()for (grouping in unique (nucmerResults_withGeneInfo_containingRibosomal$ X4)){= allMummerResults_intersectedWithNucmer_mod_withSum %>% filter (grouping == group)= pf3d7Genes %>% filter (col.0 == conservedInfo_current$ chrom[1 ], .1 >= conservedInfo_current$ chrom1_section_start[1 ] | col.2 >= conservedInfo_current$ chrom1_section_start[1 ], .1 <= conservedInfo_current$ chrom1_section_end[1 ] | col.2 <= conservedInfo_current$ chrom1_section_end[1 ])= pf3d7Genes %>% filter (col.0 == conservedInfo_current$ other_chrom[1 ], .1 >= conservedInfo_current$ chrom2_section_start[1 ] | col.2 >= conservedInfo_current$ chrom2_section_start[1 ], .1 <= conservedInfo_current$ chrom2_section_end[1 ] | col.2 <= conservedInfo_current$ chrom2_section_end[1 ])= pf3d7Chroms %>% filter (chrom %in% unique (c (conservedInfo_current$ chrom, conservedInfo_current$ other_chrom)))= pf3d7Chroms_filt %>% mutate (chrom = factor (chrom, levels = c (.$ chrom)))= conservedInfo_current %>% group_by (chrom1_section_chrom, chrom1_section_start, chrom1_section_end, %>% summarise (total = n (), revCompSum = sum (strand == "-" )) %>% mutate (chrom1RevCompFrac = revCompSum/ total) %>% mutate (chrom1RevComp = ifelse (chrom1RevCompFrac > 0.5 , T, F))= conservedInfo_current_sum %>% mutate (chrom1_section_chrom = factor (chrom1_section_chrom, levels = c (pf3d7Chroms_filt$ chrom))) %>% mutate (chrom2_section_chrom = factor (chrom2_section_chrom, levels = c (pf3d7Chroms_filt$ chrom)))#cat(c(grouping, unique(conservedInfo_current$chrom1_section_len)), sep = "\n") = ggplot (conservedInfo_current) + geom_segment (aes (x = start,xend = end,y = ifelse (strand == "-" , other_end, other_start),yend = ifelse (strand == "-" , other_start, other_end),color = strand+ geom_rect (data = pf3d7Genes_chrom1, aes (xmin = col.1 , xmax = col.2 , ymax = conservedInfo_current$ chrom2_section_start[1 ] - conservedInfo_current$ chrom2_section_len[1 ] * 0.001 , ymin = conservedInfo_current$ chrom2_section_start[1 ] - conservedInfo_current$ chrom2_section_len[1 ] * 0.01 , fill = description, ID = ID)) + geom_rect (data = pf3d7Genes_chrom2, aes (ymin = col.1 , ymax = col.2 , xmax = conservedInfo_current$ chrom1_section_start[1 ] - conservedInfo_current$ chrom1_section_len[1 ] * 0.001 , xmin = conservedInfo_current$ chrom1_section_start[1 ] - conservedInfo_current$ chrom1_section_len[1 ] * 0.01 , fill = description, ID = ID )) + labs (title = grouping, y = pf3d7Genes_chrom2$ col.0 [1 ], x = pf3d7Genes_chrom1$ col.0 [1 ]) + sofonias_theme + coord_equal () + scale_color_manual (values = c ("-" = "#AA0A3C" , "+" = "#0AB45A" ));if (conservedInfo_current_sum$ chrom1RevComp[1 ]){= currentPlot + scale_y_reverse ()= ggplotly (currentPlot)paste0 (grouping, "-fullView" )]] = ggplotly (ggplot () + geom_rect (data = pf3d7Chroms_filt,aes (xmin = 0 ,xmax = length,ymin = as.numeric (chrom) - 0.4 ,ymax = as.numeric (chrom) + 0.4 fill = "grey75" + scale_y_continuous (breaks = 1 : (nrow (pf3d7Chroms_filt)),labels = levels (pf3d7Chroms_filt$ chrom)+ + geom_rect (data = conservedInfo_current_sum,aes (xmin = chrom1_section_start,xmax = chrom1_section_end,ymin = as.numeric (chrom1_section_chrom) - 0.4 ,ymax = as.numeric (chrom1_section_chrom) + 0.4 ,fill = chrom1RevComp+ geom_rect (data = conservedInfo_current_sum,aes (xmin = chrom2_section_start,xmax = chrom2_section_end,ymin = as.numeric (chrom2_section_chrom) - 0.4 ,ymax = as.numeric (chrom2_section_chrom) + 0.4 fill = "#0AB45A" + scale_fill_manual (values = c ("TRUE" = "#AA0A3C" , "FALSE" = "#0AB45A" ))``` ```{r} #| fig-column: screen #| column: screen-inset #| results: asis #| fig-width: 15 #| fig-height: 25 cat (create_tabsetOfHtmlWidgets (listOfPlots))#htmltools::tagList(listOfPlots) ``` ```{r} #| column: screen-inset = allMummerResults_intersectedWithNucmer_mod_withSum %>% filter (group %in% nucmerResults_withGeneInfo_containingRibosomal$ X4) %>% select (group, totalBases, starts_with ("chrom1_" ), starts_with ("chrom2_" )) %>% unique ()create_dt (allMummerResults_intersectedWithNucmer_mod_withSum_sel_rRNA)``` <!-- ### chr05 chr07 rRNA - Pf3D7_07_v3-1083318-1090383-for–Pf3D7_05_v3-1289161-1296225-for --> <!-- The other two intact rRNA loci within Plasmodium falciparum are of the A-type (expressed primarily while in the human host) and the duplicated/indentical region surrunding this loci contain only the rRNA and no other surrounding genes. --> <!-- ```{bash, eval = F} --> <!-- elucidator createBedRegionFromName --names Pf3D7_05_v3-1289161-1296225-for,Pf3D7_07_v3-1083318-1090383-for | elucidator getFastaWithBed --overWrite --twoBit /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.2bit --bed STDIN --out 05_07_region.fasta --> <!-- elucidator compareToRef --fasta 05_07_region.fasta --out 05_07_region_comparison --ref 05_07_region.fasta --overWrite --> <!-- ``` --> <!-- ```{r} --> <!-- comps = bind_rows( --> <!-- readr::read_tsv( --> <!-- "surroundingRegionsMaterials/interchromosomalComparison/05_07_region_comparison.txt", --> <!-- ) %>% --> <!-- mutate(region = "rRNA_chr05_chr07_region") --> <!-- ) --> <!-- create_dt(comps) --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen --> <!-- grouping = "Pf3D7_07_v3-1083318-1090383-for--Pf3D7_05_v3-1289161-1296225-for" --> <!-- conservedInfo_current = conservedInfo_mod %>% --> <!-- filter(grouping == group) --> <!-- pf3d7Genes_chrom1 = pf3d7Genes %>% --> <!-- filter(col.0 == conservedInfo_current$chrom1[1], --> <!-- col.1 >= conservedInfo_current$chrom1_section_start[1], --> <!-- col.1 <= conservedInfo_current$chrom1_section_end[1]) %>% --> <!-- filter(description != "unspecified product") --> <!-- pf3d7Genes_chrom2 = pf3d7Genes %>% --> <!-- filter(col.0 == conservedInfo_current$chrom2[1], --> <!-- col.1 >= conservedInfo_current$chrom2_section_start[1], --> <!-- col.1 <= conservedInfo_current$chrom2_section_end[1])%>% --> <!-- filter(description != "unspecified product") --> <!-- write_tsv(conservedInfo_current, "conservedInfo_between_05_and_07_sharedRegion.tsv") --> <!-- conservedInfo_current_plot = ggplot(conservedInfo_current) + --> <!-- geom_segment( --> <!-- aes( --> <!-- x = chrom1Pos_start, --> <!-- xend = crhom1Pos_end - 1, --> <!-- y = chrom2Pos, --> <!-- yend = chrom2Pos + size - 1 --> <!-- ), --> <!-- linewidth = 2.5, --> <!-- color = "black" --> <!-- ) + --> <!-- geom_rect( --> <!-- ymin = conservedInfo_current$chrom2_section_start[1], --> <!-- ymax = conservedInfo_current$chrom2_section_start[1] + conservedInfo_current$chrom2_section_len[1], --> <!-- xmin = conservedInfo_current$chrom1_section_start[1] - conservedInfo_current$chrom1_section_len[1] * 0.1, --> <!-- xmax = conservedInfo_current$chrom1_section_start[1] - conservedInfo_current$chrom1_section_len[1] * 0.001, --> <!-- fill = "grey81") + --> <!-- geom_rect( --> <!-- ymax = conservedInfo_current$chrom2_section_start[1] - conservedInfo_current$chrom2_section_len[1] * 0.001, --> <!-- ymin = conservedInfo_current$chrom2_section_start[1] - conservedInfo_current$chrom2_section_len[1] * 0.1, --> <!-- xmin = conservedInfo_current$chrom1_section_start[1], --> <!-- xmax = conservedInfo_current$chrom1_section_start[1] + conservedInfo_current$chrom1_section_len[1], --> <!-- fill = "grey81") + --> <!-- geom_rect(data = pf3d7Genes_chrom1, --> <!-- aes(xmin = col.1, xmax = col.2, --> <!-- ymax = conservedInfo_current$chrom2_section_start[1] - conservedInfo_current$chrom2_section_len[1] * 0.001, --> <!-- ymin = conservedInfo_current$chrom2_section_start[1] - conservedInfo_current$chrom2_section_len[1] * 0.1, --> <!-- fill = description)) + --> <!-- geom_rect(data = pf3d7Genes_chrom2, --> <!-- aes(ymin = col.1, ymax = col.2, --> <!-- xmax = conservedInfo_current$chrom1_section_start[1] - conservedInfo_current$chrom1_section_len[1] * 0.001, --> <!-- xmin = conservedInfo_current$chrom1_section_start[1] - conservedInfo_current$chrom1_section_len[1] * 0.1, --> <!-- fill = description)) + --> <!-- labs(title = "", --> <!-- x = "3D7 Chr07-1289161-1296225 (bp=7,064)", --> <!-- y = "3D7 Chr05-1083318-1090383 (bp=7,065)", --> <!-- fill = "") + --> <!-- sofonias_theme + --> <!-- coord_equal() + --> <!-- # scale_fill_tableau() + --> <!-- scale_fill_manual(values = c("#2271B2", "#F748A5", "#3DB7E9")) + --> <!-- guides(fill=guide_legend(ncol = 1,byrow=TRUE)) + --> <!-- theme(legend.position = c(0.375, 0.825), --> <!-- panel.border = element_blank(), --> <!-- legend.background = element_blank(), --> <!-- legend.box.background = element_rect(colour = "black")) --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen --> <!-- #| column: screen-inset-shaded --> <!-- #| fig-width: 5 --> <!-- #| fig-height: 5 --> <!-- print(conservedInfo_current_plot) --> <!-- ``` --> <!-- ```{r} --> <!-- cairo_pdf("shared_3D7chr05-3D7chr07_region.pdf", width = 5, height = 5) --> <!-- print(conservedInfo_current_plot) --> <!-- dev.off() --> <!-- ``` --> <!-- ```{r} --> <!-- #| results: asis --> <!-- #| echo: false --> <!-- cat(createDownloadLink("shared_3D7chr05-3D7chr07_region.pdf")) --> <!-- ``` -->