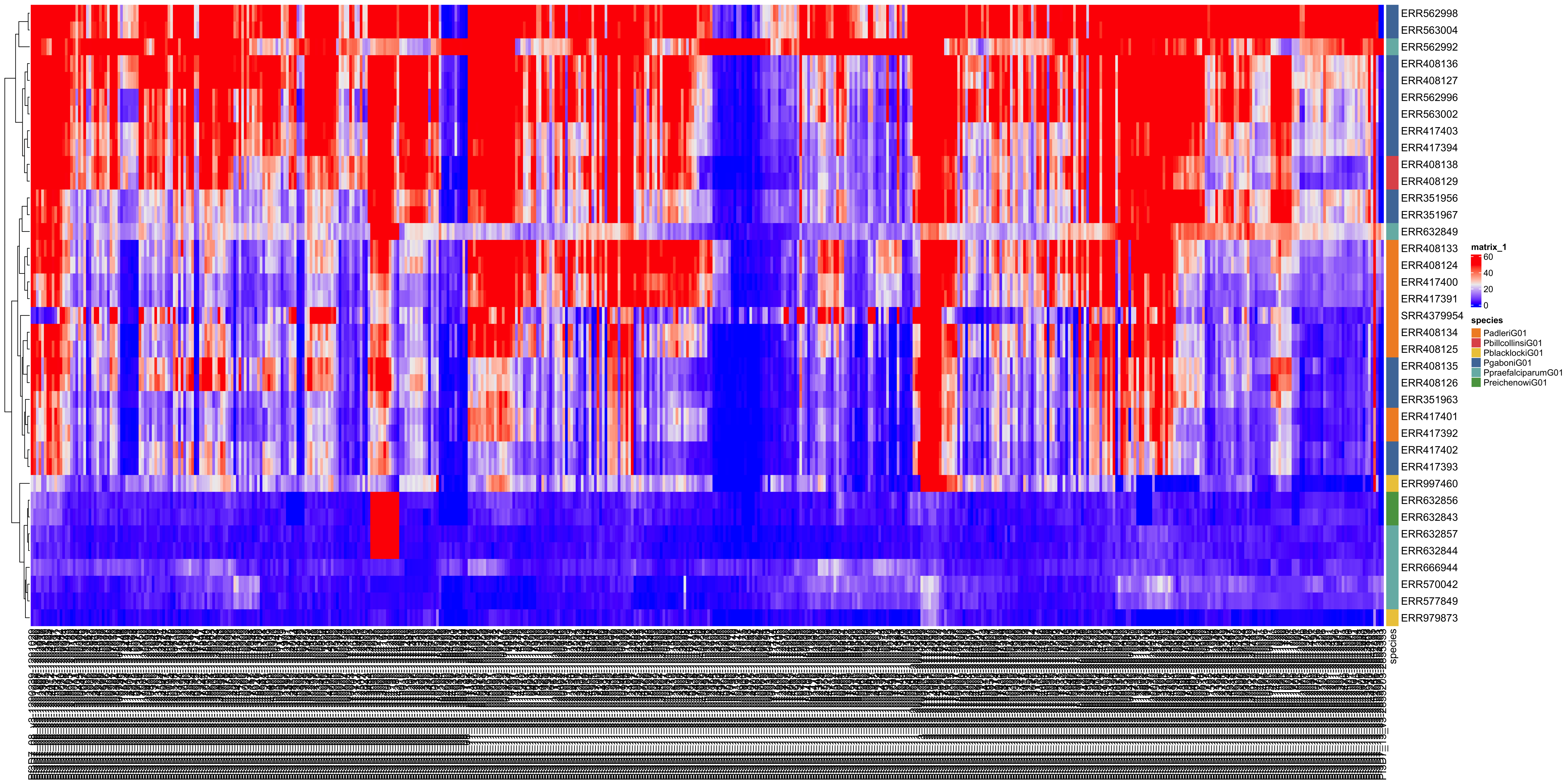
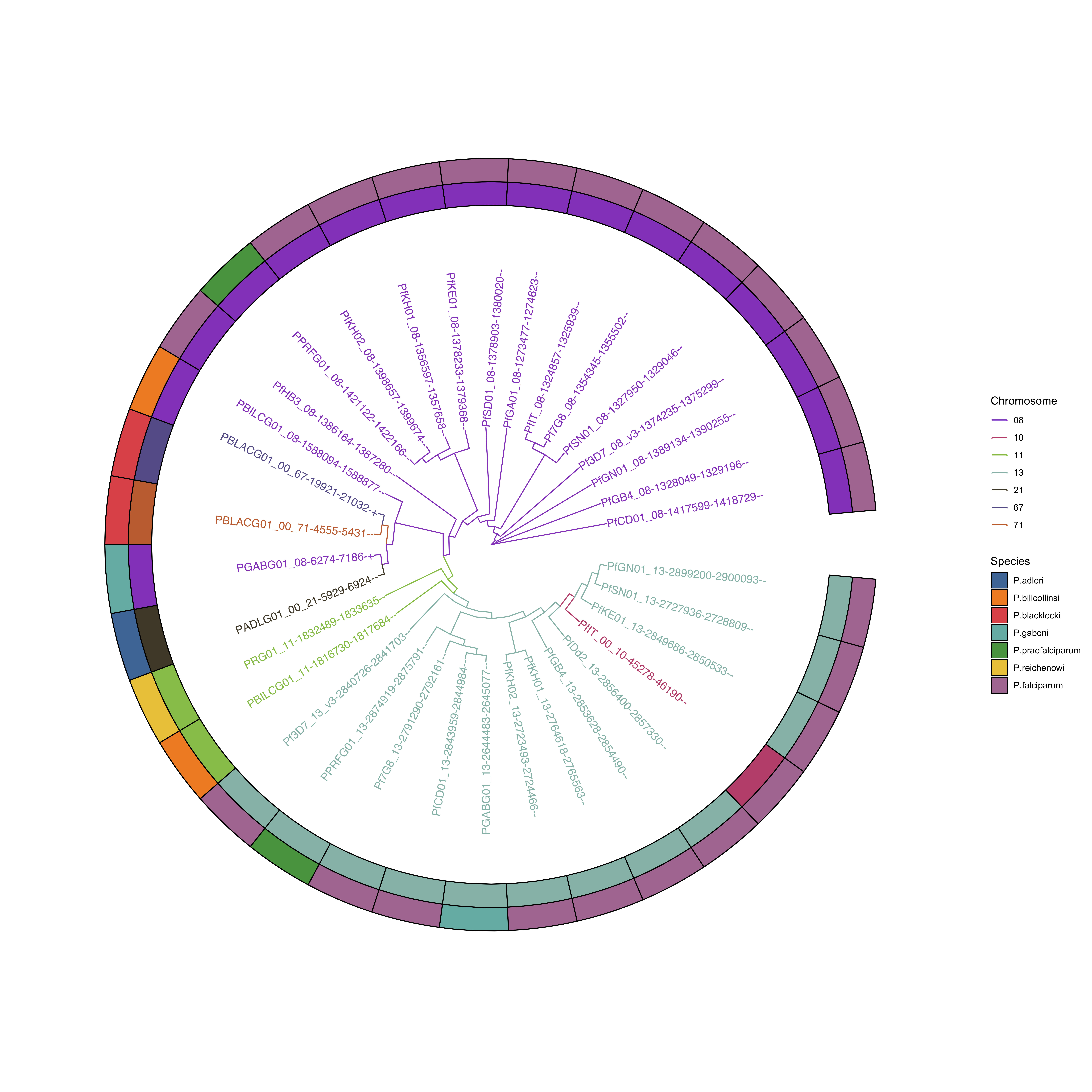
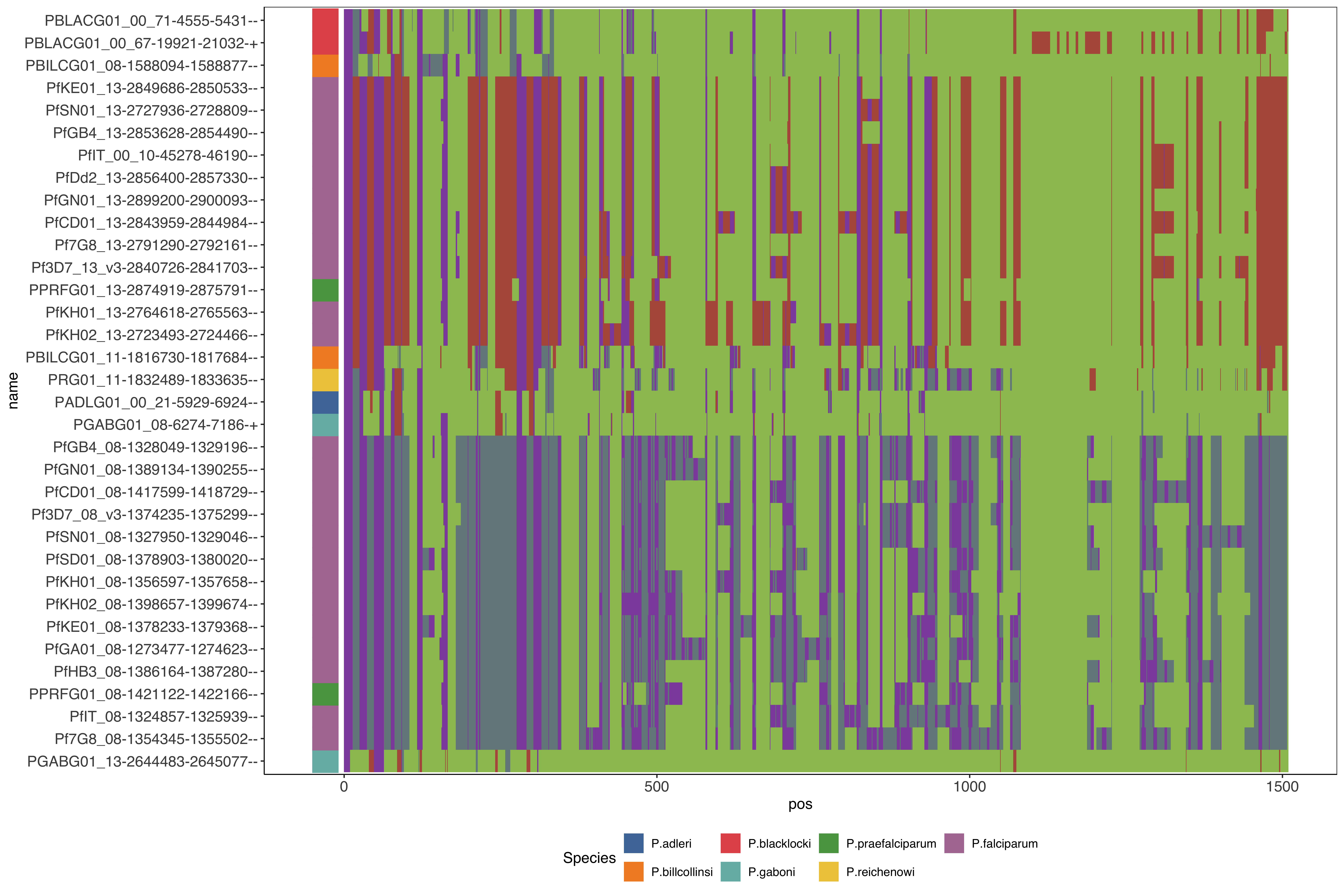
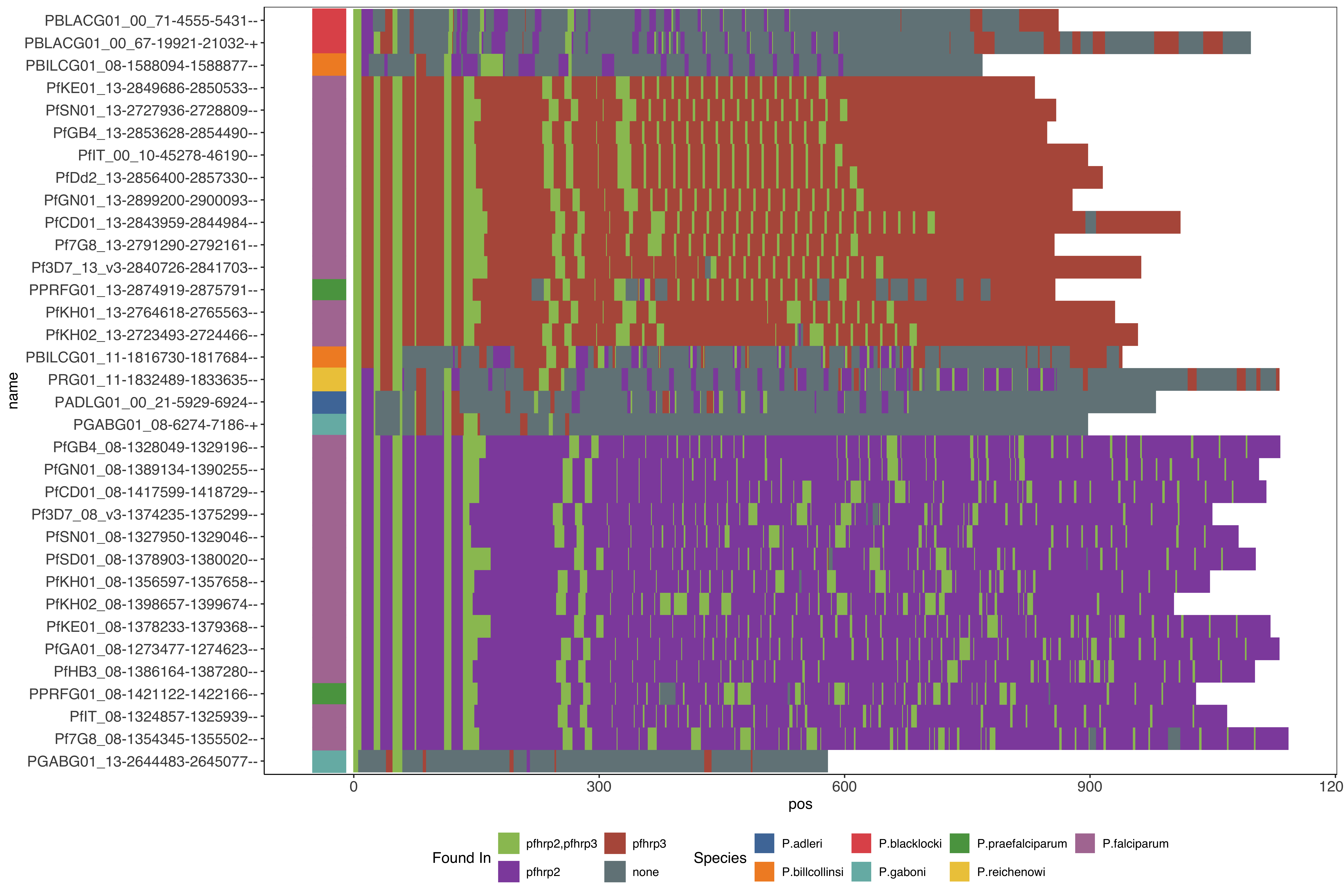
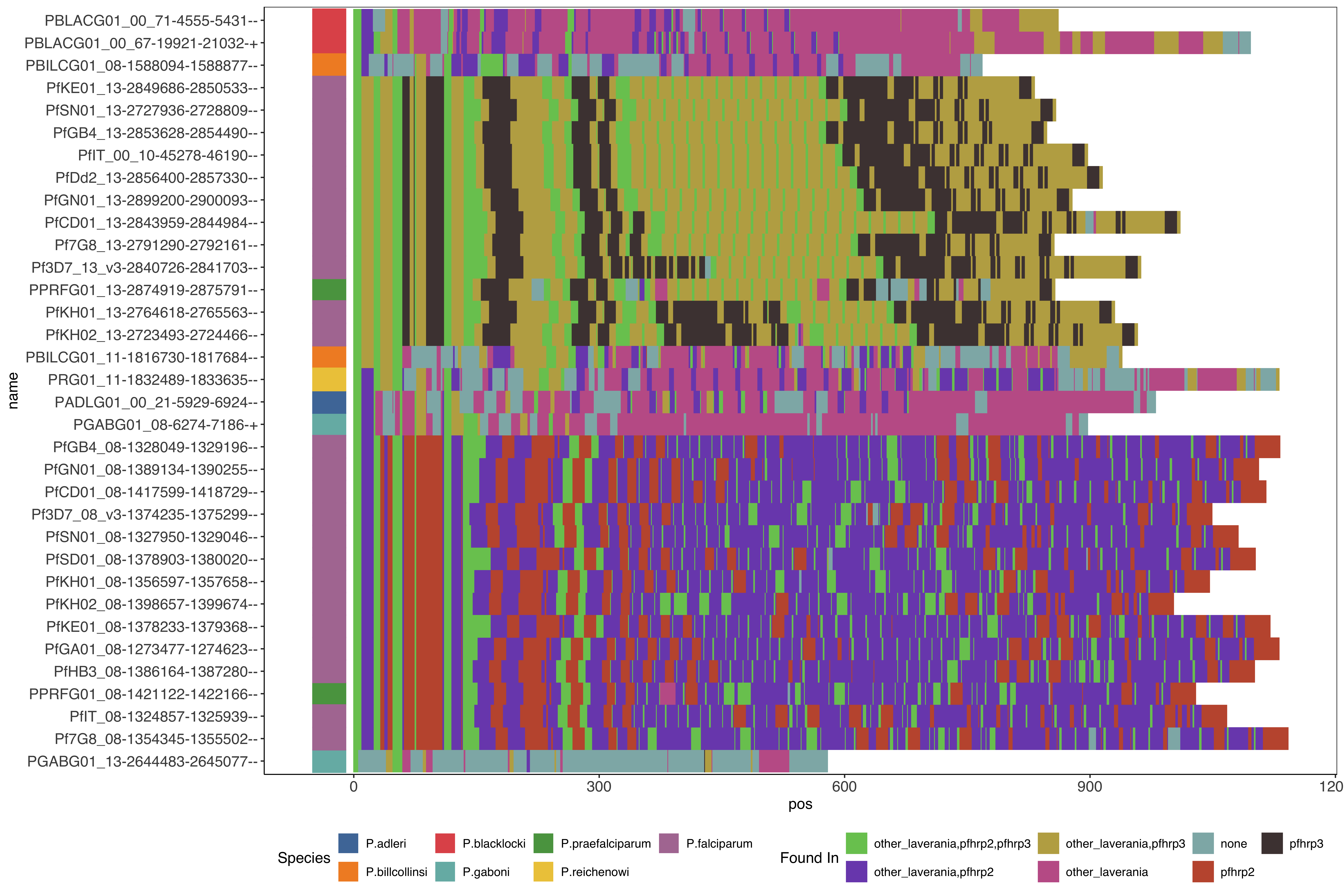
--- title: "P. laverania HRP2/HRP3 comparions" code-fold: true --- ```{r setup, echo=FALSE, message=FALSE} source("../common.R") ``` ## Getting HRP2/HRP3 regions details ```{bash, eval = F} nohup elucidator runMultipleCommands --cmdFile runRegionBamTrimCmds.txt --additionalFields "{SAMPLE}:allSamples.txt;{DIRNAME}:finalHRPII_HRPIII_windowsSubWindows;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/finalHRPII_HRPIII_windows_combinedVarConservedRegions.bed\"" --numThreads 12 & cd finalHRPII_HRPIII_windowsSubWindows PathWeaver runProcessClustersOnRecon --alnInfoDir alnCache --swgaSampleClusErrorSet --overWriteDir --fracCutOff 0.005 --dout popClusteringSWGAErrorsPartials --addPartial --oneSampOnlyOneOffHapsFrac 0.21 --removeOneSampOnlyOneOffHaps --replicateMinTotalReadCutOff 10 --clusterCutOff 5 --pat _finalHRPII_HRPIII_windowsSubWindows --numThreads 5 --rescueExcludedOneOffLowFreqHaplotypes --excludeLowFreqOneOffs --excludeCommonlyLowFreqHaplotypes nohup elucidator runMultipleCommands --cmdFile runRegionBamTrimCmds.txt --additionalFields "{SAMPLE}:allSamples.txt;{DIRNAME}:finalHRPII_HRPIII_windows;{BEDFNP}:\"beds/hrps/redesign_2020_11_22/finalHRPII_HRPIII_windows_withGeneInfo.bed\"" --numThreads 12 & cd finalHRPII_HRPIII_windows PathWeaver runProcessClustersOnRecon --alnInfoDir alnCache --swgaSampleClusErrorSet --overWriteDir --fracCutOff 0.005 --dout popClusteringSWGAErrorsPartials --addPartial --oneSampOnlyOneOffHapsFrac 0.21 --removeOneSampOnlyOneOffHaps --replicateMinTotalReadCutOff 10 --clusterCutOff 5 --pat _finalHRPII_HRPIII_windows --numThreads 5 --rescueExcludedOneOffLowFreqHaplotypes --excludeLowFreqOneOffs --excludeCommonlyLowFreqHaplotypes ``` ```{r} = readr:: read_tsv ("mashes_winners.tab.txt" )= readr:: read_tsv ("data/finalHRPII_HRPIII_windows/reports/allBasicInfo.tab.txt.gz" )= finalHRPII_HRPIII_windowsSubWindows_basicInfo %>% mutate (genomicID = paste0 (` #chrom ` , "-" , start, "-" , end)) %>% select (genomicID, sample, perBaseCoverage) %>% spread (genomicID, perBaseCoverage)= as.matrix (finalHRPII_HRPIII_windowsSubWindows_basicInfo_covSp[,2 : ncol (finalHRPII_HRPIII_windowsSubWindows_basicInfo_covSp)])rownames (finalHRPII_HRPIII_windowsSubWindows_basicInfo_covSp_mat) = finalHRPII_HRPIII_windowsSubWindows_basicInfo_covSp$ sample> 50 ] = 50 = mashWinners[match (rownames (finalHRPII_HRPIII_windowsSubWindows_basicInfo_covSp_mat), mashWinners$ sample),]= mashWinners[, c ("species" )] %>% as.data.frame ()= list ()"species" ]] = tableau_color_pal ()(6 )names (rowAnnoColors[["species" ]]) = unique (rowAnnoDf$ species)= rowAnnotation (df = rowAnnoDf, col = rowAnnoColors)``` ```{r} #| fig-column: screen-inset-shaded #| fig-height: 12.5 #| fig-width: 25 :: Heatmap (finalHRPII_HRPIII_windowsSubWindows_basicInfo_covSp_mat, cluster_columns = F, right_annotation = rowAnno)``` ```{bash, eval = F} cd combiningCombinedHrps/finalCalls gegrep PBLAC ../../hmm_hrps_againstAllPlaverania/noOverlapMergedFiltHits.fasta -A1 --no-group-separator > laverania_from_hmm.fasta sed 's/\[/;/g' hmm_pblac.fasta | sed 's/PBL/\[chrom=PBL/g' | sed 's/revStrand=true/revStrand=-/g' | sed 's/revStrand=false/revStrand=+/g'> renamed_hmm_pblac.fasta elucidator renameSeqsWithMetaField --fasta renamed_hmm_pblac.fasta --metaFields chrom,trimStart,trimEnd,revStrand --overWrite grep PgaboniG01 -A1 ../../extractedHRPII/Pf3D7_08_v3-1374235-1375299-rev/allRefs.fasta > laverania_from_lastz.fasta grep PbillcollinsiG01 -A1 ../../extractedHRPII/Pf3D7_08_v3-1374235-1375299-rev/allRefs.fasta >> laverania_from_lastz.fasta grep PreichenowiG01 -A1 ../../extractedHRPII/Pf3D7_08_v3-1374235-1375299-rev/allRefs.fasta >> laverania_from_lastz.fasta grep PpraefalciparumG01 -A1 ../../extractedHRPII/Pf3D7_08_v3-1374235-1375299-rev/allRefs.fasta >> laverania_from_lastz.fasta grep PadleriG01 -A1 ../../extractedHRPII/Pf3D7_08_v3-1374235-1375299-rev/allRefs.fasta >> laverania_from_lastz.fasta grep PgaboniG01 -A1 ../../extractedHRPIII/Pf3D7_13_v3-2840726-2841703-rev/allRefs.fasta >> laverania_from_lastz.fasta grep PbillcollinsiG01 -A1 ../../extractedHRPIII/Pf3D7_13_v3-2840726-2841703-rev/allRefs.fasta >> laverania_from_lastz.fasta grep PreichenowiG01 -A1 ../../extractedHRPIII/Pf3D7_13_v3-2840726-2841703-rev/allRefs.fasta >> laverania_from_lastz.fasta grep PpraefalciparumG01 -A1 ../../extractedHRPIII/Pf3D7_13_v3-2840726-2841703-rev/allRefs.fasta >> laverania_from_lastz.fasta elucidator renameSeqsWithMetaField --metaFields chrom,start,end,strand --overWrite --fasta laverania_from_lastz.fasta cat renamed_renamed_hmm_pblac.fasta renamed_laverania_from_lastz.fasta > other_laverania.fasta rsync -raPh ../../../../../mappingOutSurroundingRegions/extractedHRPII/Pf3D7_08_v3-1374235-1375299-rev/allRefs.fasta pfhrp2.fasta rsync -raPh ../../../../../mappingOutSurroundingRegions/extractedHRPIII/Pf3D7_13_v3-2840726-2841703-rev/allRefs.fasta pfhrp3.fasta elucidator renameSeqsWithMetaField --metaFields chrom,start,end,strand --overWrite --fasta pfhrp2.fasta elucidator renameSeqsWithMetaField --metaFields chrom,start,end,strand --overWrite --fasta pfhrp3.fasta cat other_laverania.fasta renamed_pfhrp2.fasta renamed_pfhrp3.fasta > all_laverania.fasta muscle -in all_laverania.fasta -out aln_all_laverania.fasta elucidator removeBestSubSeq --fasta all_laverania.fasta --ref intron.fasta --overWrite --out cdna_all_laverania.fasta; # some mods for PADLG01_00_21-5929-6924 to fix probable erroneous stop codonds internal sed 's/CTGCTCCCATGCAG/CTGCTCACCATGCAG/g' cdna_all_laverania.fasta > mod.fasta; sed 's/ATGCTCACTCATGCAGGT/ATGCTCATCATGCAGGT/g' mod.fasta > mod2.fasta; sed 's/CATGCAGGTGCTGCTCTGCTCACC/CATGCAGGTGCTGCTCATCATGCTCACC/g' mod2.fasta > mod3.fasta elucidator guessAProteinFromSeq --fasta mod3.fasta --overWrite --out translated_cdna_all_laverania.fasta --getLongest --removeTrailingStop muscle -in translated_cdna_all_laverania.fasta -out aln_translated_cdna_all_laverania.fasta FastTree aln_translated_cdna_all_laverania.fasta > translated_cdna_all_laverania.nwk ``` ```{r, fig.width=15, fig.height=15} library(ggtree) library(ggtreeExtra) hrps_tree = ape::read.tree("combiningCombinedHrps/finalCalls/translated_cdna_all_laverania.nwk") hrps_tree_labelDf = tibble(seq = hrps_tree$tip.label) %>% mutate(chromNum = gsub("-.*", "", seq)) %>% mutate(chromNum = gsub("_v3", "", chromNum)) %>% mutate(chromNum = gsub(".*_", "", chromNum)) %>% mutate(genome = gsub("_.*", "", seq)) %>% separate(seq, into = c("chrom", "start", "end"), sep = "-", convert = T, remove = F) %>% arrange(genome, chrom, start) %>% group_by(genome, chromNum) %>% mutate(chromID = row_number(), total = n()) %>% mutate(chromLabel = ifelse(total > 1, paste0(chromNum, "-", chromID), chromNum ) ) chroms = list() for(currentChrom in unique(hrps_tree_labelDf$chromNum)){ hrps_tree_labelDf_chrom = hrps_tree_labelDf %>% filter(chromNum == currentChrom) chroms[[currentChrom]] = hrps_tree_labelDf_chrom$seq } hrps_tree = groupOTU(hrps_tree, chroms) ``` ```{r, fig.width=15, fig.height=15} #| fig-column: screen hrps_tree_labelDf = hrps_tree_labelDf %>% mutate(strainSuffix = substr(seq, 1,ifelse(grepl("Pf", seq), 2, 3)))%>% mutate(strainSuffix = case_when( strainSuffix == "Pf" ~ "P.falciparum", strainSuffix == "PAD" ~ "P.adleri", strainSuffix == "PBI" ~ "P.billcollinsi", strainSuffix == "PBL" ~ "P.blacklocki", strainSuffix == "PGA" ~ "P.gaboni", strainSuffix == "PPR" ~ "P.praefalciparum", strainSuffix == "PRG" ~ "P.reichenowi" )) hrps_tree_labelDf_forAnnotation = hrps_tree_labelDf %>% group_by() %>% select(seq, strainSuffix, chromNum) %>% gather(annotate, value, 2:3) strainFills = unique(hrps_tree_labelDf$strainSuffix) strainFills = tableau_color_pal()(length(strainFills)) names(strainFills) = unique(hrps_tree_labelDf$strainSuffix) chromosomeColors = scheme$hex(length(chroms)) names(chromosomeColors) = names(chroms) hrps_tree_plot = ggtree(hrps_tree, layout = 'fan', branch.length = 'none', aes(color = group)) + geom_tiplab() + scale_color_manual("Chromosome", values = chromosomeColors, guide = guide_legend(override.aes = list(shape = 1, size = 1)) ) + hexpand(0.5) + #theme(plot.margin = margin(120, 120, 120, 120)) + scale_fill_manual( "Species", values = c(strainFills, chromosomeColors), breaks = names(strainFills), labels = names(strainFills) ) print(hrps_tree_plot + geom_fruit(data=hrps_tree_labelDf_forAnnotation, geom=geom_tile, mapping=aes(y=seq, x=annotate, fill=value), color = "black", offset = 1,size = 0.5) ) ``` ```{r} pdf ("hrps_tree_laverania.pdf" , width = 15 , height = 15 , useDingbats = F)print (hrps_tree_plot + geom_fruit (data= hrps_tree_labelDf_forAnnotation, geom= geom_tile,mapping= aes (y= seq, x= annotate, fill= value),color = "black" , offset = 1.1 ,size = 0.5 ) )dev.off ()``` ```{bash, eval = F} cd combiningCombinedHrps elucidator findKmersInSets --fasta unique_combinedLavPlusAllPf.fasta --seqSetFnps ../../../../mappingOutSurroundingRegions/extractedHRPII/Pf3D7_08_v3-1374235-1375299-rev/hrp2.fasta,../../../../mappingOutSurroundingRegions/extractedHRPII/Pf3D7_08_v3-1374235-1375299-rev/hrp3.fasta --dout countingKmersInPfHrp2Hrp3_k15 --kmerLength 15 --overWriteDir elucidator findKmersInSets --fasta unique_combinedLavPlusAllPf.fasta --seqSetFnps ../../../../mappingOutSurroundingRegions/extractedHRPII/Pf3D7_08_v3-1374235-1375299-rev/hrp2.fasta,../../../../mappingOutSurroundingRegions/extractedHRPII/Pf3D7_08_v3-1374235-1375299-rev/hrp3.fasta,combinedJustLav.fasta --minOccurrences 3 --dout countingKmersInPfHrp2Hrp3_k15 --kmerLength 15 --overWriteDir ``` ```{bash, eval = F} cd combiningCombinedHrps/finalCalls elucidator findKmersInSets --fasta all_laverania.fasta --seqSetFnps pfhrp2.fasta,pfhrp3.fasta --minOccurrences 2 --dout countingKmersInPfHrp2Hrp3_k15 --kmerLength 15 --overWriteDir elucidator findKmersInSets --fasta all_laverania.fasta --seqSetFnps pfhrp2.fasta,pfhrp3.fasta,other_laverania.fasta --minOccurrences 2 --dout countingKmersInPfHrp2Hrp3_plusLavs_k15 --kmerLength 15 --overWriteDir elucidator findKmersInSets --fasta aln_all_laverania.fasta --seqSetFnps pfhrp2.fasta,pfhrp3.fasta --minOccurrences 2 --dout aln_countingKmersInPfHrp2Hrp3_k11 --kmerLength 11 --overWriteDir ``` ```{r} #| fig-column: screen #| fig-width: 15 #| fig-height: 10 = readr:: read_tsv ("combiningCombinedHrps/finalCalls/aln_countingKmersInPfHrp2Hrp3_k11/seqsKmerClassified.tab.txt.gz" ) %>% mutate (name = factor (name, levels = unique (.$ name)))= kmersClassified_maln %>% select (name) %>% unique () %>% mutate (strainSuffix = substr (name, 1 ,ifelse (grepl ("Pf" , name), 2 , 3 ))) %>% mutate (strainSuffix = case_when (== "Pf" ~ "P.falciparum" , == "PAD" ~ "P.adleri" , == "PBI" ~ "P.billcollinsi" , == "PBL" ~ "P.blacklocki" , == "PGA" ~ "P.gaboni" , == "PPR" ~ "P.praefalciparum" , == "PRG" ~ "P.reichenowi" = tableau_color_pal (palette = "Superfishel Stone" )(length (unique (kmersClassified_maln$ foundIn)))= scheme$ hex (length (unique (kmersClassified_maln$ foundIn)))names (presenceColors) = unique (kmersClassified_maln$ foundIn)ggplot (kmersClassified_maln) + geom_tile (aes (x = pos, y = name, fill = foundIn)) + + geom_rect (aes (xmin = - 50 , xmax = - 10 , ymin = as.numeric (name) - 0.5 , ymax = as.numeric (name) + 0.5 , fill = strainSuffix, color = strainSuffix),data = kmersClassified_maln_names+ scale_fill_manual ("Species" ,values = c (presenceColors, strainFills),breaks = names (strainFills),labels = names (strainFills)+ scale_color_manual ("Found In" ,values = c (presenceColors, strainFills),breaks = names (presenceColors),labels = names (presenceColors),guide = guide_legend (override.aes = list (color = presenceColors,fill = presenceColors,alpha = 1 ,shape = 22 ,size = 5 nrow = 2 ``` ```{r} #| fig-column: screen #| fig-width: 15 #| fig-height: 10 = readr:: read_tsv ("combiningCombinedHrps/finalCalls/countingKmersInPfHrp2Hrp3_k15/seqsKmerClassified.tab.txt.gz" ) %>% mutate (name = factor (name, levels = unique (kmersClassified_maln$ name)))= kmersClassified %>% select (name) %>% unique () %>% mutate (strainSuffix = substr (name, 1 ,ifelse (grepl ("Pf" , name), 2 , 3 ))) %>% mutate (strainSuffix = case_when (== "Pf" ~ "P.falciparum" , == "PAD" ~ "P.adleri" , == "PBI" ~ "P.billcollinsi" , == "PBL" ~ "P.blacklocki" , == "PGA" ~ "P.gaboni" , == "PPR" ~ "P.praefalciparum" , == "PRG" ~ "P.reichenowi" = tableau_color_pal (palette = "Superfishel Stone" )(length (unique (kmersClassified$ foundIn)))= scheme$ hex (length (unique (kmersClassified$ foundIn)))names (presenceColors) = unique (kmersClassified$ foundIn)= ggplot (kmersClassified) + geom_tile (aes (x = pos, y = name, fill = foundIn, color = foundIn)) + + geom_rect (aes (xmin = - 50 , xmax = - 10 , ymin = as.numeric (name) - 0.5 , ymax = as.numeric (name) + 0.5 , fill = strainSuffix, color = strainSuffix),data = kmersClassified_names+ scale_fill_manual ("Species" ,values = c (presenceColors, strainFills),breaks = names (strainFills),labels = names (strainFills)+ scale_color_manual ("Found In" ,values = c (presenceColors, strainFills),breaks = names (presenceColors),labels = names (presenceColors),guide = guide_legend (override.aes = list (color = presenceColors,fill = presenceColors,alpha = 1 ,shape = 22 ,size = 5 nrow = 2 print (hrps_kmersClassified_plot)``` ```{r} pdf ("hrps_kmersClassified_plot.pdf" , width = 15 , height = 15 , useDingbats = F)print (hrps_kmersClassified_plot)dev.off ()``` ```{r} #| fig-column: screen #| fig-width: 15 #| fig-height: 10 = readr:: read_tsv ("combiningCombinedHrps/finalCalls/countingKmersInPfHrp2Hrp3_plusLavs_k15/seqsKmerClassified.tab.txt.gz" ) %>% mutate (name = factor (name, levels = unique (kmersClassified_maln$ name)))= kmersClassified %>% select (name) %>% unique () %>% mutate (strainSuffix = substr (name, 1 ,ifelse (grepl ("Pf" , name), 2 , 3 ))) %>% mutate (strainSuffix = case_when (== "Pf" ~ "P.falciparum" , == "PAD" ~ "P.adleri" , == "PBI" ~ "P.billcollinsi" , == "PBL" ~ "P.blacklocki" , == "PGA" ~ "P.gaboni" , == "PPR" ~ "P.praefalciparum" , == "PRG" ~ "P.reichenowi" = tableau_color_pal (palette = "Superfishel Stone" )(length (unique (kmersClassified$ foundIn)))= scheme$ hex (length (unique (kmersClassified$ foundIn)))names (presenceColors) = unique (kmersClassified$ foundIn)= ggplot (kmersClassified) + geom_tile (aes (x = pos, y = name, fill = foundIn, color = foundIn)) + + geom_rect (aes (xmin = - 50 , xmax = - 10 , ymin = as.numeric (name) - 0.5 , ymax = as.numeric (name) + 0.5 , fill = strainSuffix, color = strainSuffix),data = kmersClassified_names+ scale_fill_manual ("Species" ,values = c (presenceColors, strainFills),breaks = names (strainFills),labels = names (strainFills)+ scale_color_manual ("Found In" ,values = c (presenceColors, strainFills),breaks = names (presenceColors),labels = names (presenceColors),guide = guide_legend (override.aes = list (color = presenceColors,fill = presenceColors,alpha = 1 ,shape = 22 ,size = 5 nrow = 2 print (hrps_kmersClassified_plot)``` # All by all comparison ```{bash, eval = F} elucidator compareAllByAll --fasta all_laverania.fasta --alnInfoDir alnCache --numThreads 15 --verbose --out allByAll_all_laverania.tab.txt ``` ```{r} = readr:: read_tsv ("combiningCombinedHrps/finalCalls/allByAll_all_laverania.tab.txt" )create_dt (allByAll_all_laverania)``` ## Best hits against 3D7 ```{r} = allByAll_all_laverania %>% filter (grepl ("Pf3D7" , OtherReadId) & ! grepl ("^Pf" , ReadId)) %>% group_by (ReadId) %>% arrange (desc (perId)) %>% mutate (matchID = row_number ())= allByAll_all_laverania_against3D7 %>% filter (matchID == 1 )create_dt (allByAll_all_laverania_against3D7_best)```