Processing Samples with HRP2 Deletions TARE1
Set up
Downloads
Reading in data
Code
allMetaSamplesByBio = readr::read_tsv("../../meta/metadata/metaByBioSample.tab.txt")
allMetaSamples = readr::read_tsv("../../meta/metadata/meta.tab.txt")
allMetaDeletionCalls = readr::read_tsv("../metaSelected.tab.txt")
allMetaDeletionCalls_possiblyHRP2Deleted = allMetaDeletionCalls %>%
filter(possiblyHRP2Deleted)
allMetaDeletionCalls_possiblyChr11Deleted = allMetaDeletionCalls %>%
filter(possiblyChr11Deleted)
allMetaDeletionCalls_Hrp3pattern2 = readr::read_tsv("../allMetaDeletionCalls_Hrp3pattern2.tab.txt")
finalHRPII_HRPIII_windows = readr::read_tsv("../../windowAnalysis/windows/finalHRPII_HRPIII_windows_withTuned.bed", col_names = F) %>%
mutate(genomicID = paste0(X1, "-", X2, "-", X3)) %>%
mutate(chrom = X1, len = X3 -X2) %>%
rename(name = X4)
realmccoilCoiCalls = readr::read_tsv("../wgs_variants/THEREALMcCOIL/categorical_method/real_mccoil_COI_calls.tsv")
realmccoilCoiCalls_poly = realmccoilCoiCalls %>%
filter(random_median != 1 | topHE_median != 1) %>%
left_join(allMetaSamples %>%
select(sample, BiologicalSample))HRP2
Getting regions with TARE1 sequence
Code
nohup elucidator runMultipleCommands --cmdFile runRegionCmds.txt --additionalFields "{DIRNAME}:out_rna_PF3D7_0831800-1;{SAMPLE}:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_possiblyHRP2Deleted_samples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/hrps/out_rna_PF3D7_0831800-1.bed\";{NCPUS}:4 --keepTemporaryFiles --qualCheck 5 --keepImproperMates" --replaceFields --numThreads 11 &
cd out_rna_PF3D7_0831800-1
elucidator countPWExtractedReadsWithPattern --seqPat "TT[CT]AGGGTT[CT]AGGG","CCCT[AG]AACCCT[AG]AA" --pat _out_rna_PF3D7_0831800-1 --bedFnp /tank/data/plasmodium/falciparum/beds/hrps/out_rna_PF3D7_0831800-1.bed --minReadCounts 1 --numThreads 10 --overWrite --out out_rna_PF3D7_0831800-1_countTares.txt --samples /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_possiblyHRP2Deleted_samples.txt
cat */partial/all*.fasta */final/all*.fasta > allCombined.fasta
egrep "CCCT[AG]AACCCT[AG]AA" *_out_rna_PF3D7_0831800-1/*/*_extraction/*.fastq | sed 's/_.*//g' | sort | uniqSamples with TARE1
D10-UCSF
PP0002-C
PP0011-C
PP0017-C
PP0024-C
PP0025-C
PP0026-C
PP0028-C
PP0029-C
Code
nohup elucidator runMultipleCommands --cmdFile runRegionCmds.txt --additionalFields "{DIRNAME}:PF3D7_0831700;{SAMPLE}:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_possiblyHRP2Deleted_samples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/allGenes/PF3D7_0831700.bed\";{NCPUS}:4 --keepTemporaryFiles --qualCheck 5 --keepImproperMates" --replaceFields --numThreads 11 &
egrep "CCCT[AG]AACCCT[AG]AA" */*/*_extraction/*.fastq | sed 's/_.*//g' | sort | uniqPfDd2
Code
finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full = finalHRPII_HRPIII_windows %>%
filter("Pf3D7_08_v3" == X1, X2 >= 1365360, X3 <= 1375435)
finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out = finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full %>%
group_by(X1) %>% summarise(X2 = min(X2),
X3 = max(X3)) %>%
mutate(name = paste0(X1, "-", X2, "-", X3)) %>%
mutate(len = X3 - X2,
strand = "+")
write_tsv(finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out %>%
select(1:6), "finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out.bed", col_names = F)
write_tsv(finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full %>%
select(1:6), "finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select.bed", col_names = F)Code
elucidator createWindowsInRegions --bed finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select.bed --step 50 --windowSize 100 --minLen 50 | elucidator bedGetIntersectingGenesInGff --gff /tank/data/genomes/plasmodium/genomes/pf/info/gff/Pf3D7.gff --extraAttributes description --bed STDIN --overWrite --out finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select_step50_windowSize100.bed
rsync -raPh finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select_step50_windowSize100.bed hathawan@calderon.barrel-of-knowledge.info:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22building file list ...
0 files...
1 file to consider
finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select_step50_windowSize100.bed
700 4% 0.00kB/s 0:00:00
15.62K 100% 14.23MB/s 0:00:00 (xfer#1, to-check=0/1)
sent 290 bytes received 180 bytes 313.33 bytes/sec
total size is 15.62K speedup is 33.24Code
nohup elucidator runMultipleCommands --cmdFile runRegionCmds.txt --additionalFields "{DIRNAME}:finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select_step50_windowSize100_NoTrim;{SAMPLE}:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_possiblyHRP2Deleted_samples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select_step50_windowSize100.bed\";{NCPUS}:4 --keepTemporaryFiles --qualCheck 5 --keepImproperMates" --replaceFields --numThreads 11 &
cd finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select_step50_windowSize100_NoTrim
elucidator countPWExtractedReadsWithPattern --seqPat "TT[CT]AGGGTT[CT]AGGG","CCCT[AG]AACCCT[AG]AA" --pat _finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select_step50_windowSize100_NoTrim --bedFnp /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select_step50_windowSize100.bed --minReadCounts 1 --numThreads 10 --overWrite --out finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select_step50_windowSize100_countTares.txt --samples /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_possiblyHRP2Deleted_samples.txt
PathWeaver runProcessClustersOnRecon --overWriteDir --dout popClustering --pat _finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select_step50_windowSize100_NoTrim --numThreads 5 --groupingsFile /tank/data/plasmodium/falciparum/pfdata/metadata/meta.tab.txt --samples /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_possiblyHRP2Deleted_samples.txtCode
elucidator createWindowsInRegions --bed finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out.bed --step 50 --windowSize 100 --minLen 50 | elucidator bedGetIntersectingGenesInGff --gff /tank/data/genomes/plasmodium/genomes/pf/info/gff/Pf3D7.gff --extraAttributes description --bed STDIN --overWrite --out finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100.bed
rsync -raPh finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100.bed hathawan@calderon.barrel-of-knowledge.info:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22building file list ...
0 files...
1 file to consider
finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100.bed
700 2% 0.00kB/s 0:00:00
24.66K 100% 22.85MB/s 0:00:00 (xfer#1, to-check=0/1)
sent 344 bytes received 258 bytes 401.33 bytes/sec
total size is 24.66K speedup is 40.97Code
nohup elucidator runMultipleCommands --cmdFile runRegionCmds.txt --additionalFields "{DIRNAME}:finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100_NoTrim;{SAMPLE}:/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_possiblyHRP2Deleted_samples.txt;{BEDFNP}:\"/tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100.bed\";{NCPUS}:4 --keepTemporaryFiles --qualCheck 5 --keepImproperMates" --replaceFields --numThreads 11 &
cd finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100_NoTrim
elucidator countPWExtractedReadsWithPattern --seqPat "TT[CT]AGGGTT[CT]AGGG","CCCT[AG]AACCCT[AG]AA" --pat _finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100_NoTrim --bedFnp /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100.bed --minReadCounts 1 --numThreads 10 --overWrite --out finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100_countTares.txt --samples /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_possiblyHRP2Deleted_samples.txt
PathWeaver runProcessClustersOnRecon --overWriteDir --dout popClustering --pat _finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100_NoTrim --numThreads 5 --groupingsFile /tank/data/plasmodium/falciparum/pfdata/metadata/meta.tab.txt --samples /tank/data/plasmodium/falciparum/beds/hrps/redesign_2020_11_22/allMetaDeletionCalls_possiblyHRP2Deleted_samples.txtCode
hrp2Del_samplesWithTare1 = tibble(
sample = c(
"D10-UCSF",
"IGS-BRA-004sA",
"IGS-CBD-031",
"PfDd2",
"PP0002-C",
"PP0011-C",
"PP0017-C",
"PP0024-C",
"PP0025-C",
"PP0026-C",
"PP0028-C",
"PP0029-C"
)
)
hrp2Del_samplesWithTare1 = hrp2Del_samplesWithTare1 %>%
left_join(allMetaDeletionCalls)
create_dt(hrp2Del_samplesWithTare1)Plotting Coverage
Downloads
Code
cov = readr::read_tsv("../../meta/allCov_summaryStats.tab.txt.gz")
allReports = readr::read_tsv("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_select_step50_windowSize100_NoTrim/popClustering/reports/allBasicInfo.tab.txt.gz") %>%
filter(sample %!in% realmccoilCoiCalls_poly$sample) %>%
mutate(inGene = !is.na(extraField0)) %>%
left_join(cov) %>%
mutate(medianCov = median(perBaseCoverage),
meanCov = mean(perBaseCoverage)) %>%
mutate(perBaseCoverageNorm = ifelse(inGene, perBaseCoverage/medianPerBaseCov_inGenes, perBaseCoverage/medianPerBaseCov_notInGenes)) %>%
#mutate(perBaseCoverageNorm = ifelse(inGene, perBaseCoverage/meanPerBaseCov_inGenes, perBaseCoverage/meanPerBaseCov_notInGenes)) %>%
mutate(perBaseCoverageNormRounded = ifelse(perBaseCoverageNorm >0.10 & perBaseCoverageNorm <= 1, 1, perBaseCoverageNorm)) %>%
mutate(perBaseCoverageNormRounded = round(perBaseCoverageNormRounded)) %>%
mutate(perBaseCoverageNormRounded = ifelse(grepl("PP", sample) & perBaseCoverageNormRounded > 1, 1, perBaseCoverageNormRounded))
allReports_sp = allReports %>%
select(sample, name, perBaseCoverageNormRounded) %>%
spread(name, perBaseCoverageNormRounded)
allReports_sp_mat = as.matrix(allReports_sp[,2:ncol(allReports_sp)])
rownames(allReports_sp_mat) = allReports_sp$sample
allReports_sp_mat[allReports_sp_mat >2] = 2
annotationTextSize = 20
topAnno = allReports %>%
filter(sample == .$sample[1]) %>%
select(name, extraField0) %>%
rename(`Gene Description` = extraField0) %>%
mutate(`Gene Description` = gsub(".*description=", "", `Gene Description`))%>%
mutate(`Gene Description` = gsub("\\].*", "", `Gene Description`)) %>%
mutate(`Gene Description` = ifelse(grepl("PHIST", `Gene Description`), gsub("\\).*", "", gsub(".*PHIST", "PHIST", `Gene Description`)), `Gene Description`))
topAnnoDf = topAnno %>%
select(-name) %>%
as.data.frame()
topAnnoColors = createColorListFromDf(topAnnoDf)
metaReSelected = allMetaDeletionCalls[match(rownames(allReports_sp_mat), allMetaDeletionCalls$sample), ]
rowAnno = tibble(metaReSelected[,c("sample","country")]) %>%
mutate(`TARE1 Present on Chr8` = sample %in% hrp2Del_samplesWithTare1$sample)
rowAnnoDf = rowAnno %>%
select(-sample) %>%
as.data.frame()
rowAnnoColors = createColorListFromDf(rowAnnoDf)
sideAnno = rowAnnotation(
df = rowAnnoDf,
col = rowAnnoColors,
gp = gpar(col = "grey10"),
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
gap = unit(0.1, "cm")
)
topAnno = HeatmapAnnotation(
df = topAnnoDf,
col = topAnnoColors,
annotation_name_gp = gpar(fontsize = annotationTextSize),
annotation_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold")
),
annotation_name_side = "left",
na_col = "#FFFFFF00"
)
library(circlize)
col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3)))
allReports_sp_mat_nolab = allReports_sp_mat
colnames(allReports_sp_mat_nolab) = NULL
allReports_sp_mat_hm = Heatmap(
allReports_sp_mat_nolab,
cluster_columns = F,
col = col_fun,
name = "coverage",
top_annotation = topAnno,
right_annotation = sideAnno,
row_dend_width = unit(5, "cm"),
column_dend_height = unit(5, "cm"),
heatmap_legend_param = list(
labels_gp = gpar(fontsize = annotationTextSize),
title_gp = gpar(fontsize = annotationTextSize, fontface = "bold"),
at = c(0, 0.5, 1, 1.5, 2),
labels = c("0", "0.5x", "1.0x", "1.5x", ">=2x")
),
cell_fun = function(j, i, x, y, width, height, fill) {
grid.rect(x = x, y = y, width = width, height = height *.9,
gp = gpar(fill = fill, col = NA))
},
rect_gp = gpar(type = "none"),
column_title = "Chr08[1,365kb:1,375kb]",
column_title_gp = gpar(fontsize = 20, fontface = "italic")
)Final Plot
Country and whether or not TARE1 was detected found within the right annotation bar, the top bar has the genes
Code
draw(allReports_sp_mat_hm, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", padding = unit(c(25, 2, 2, 2), "mm"))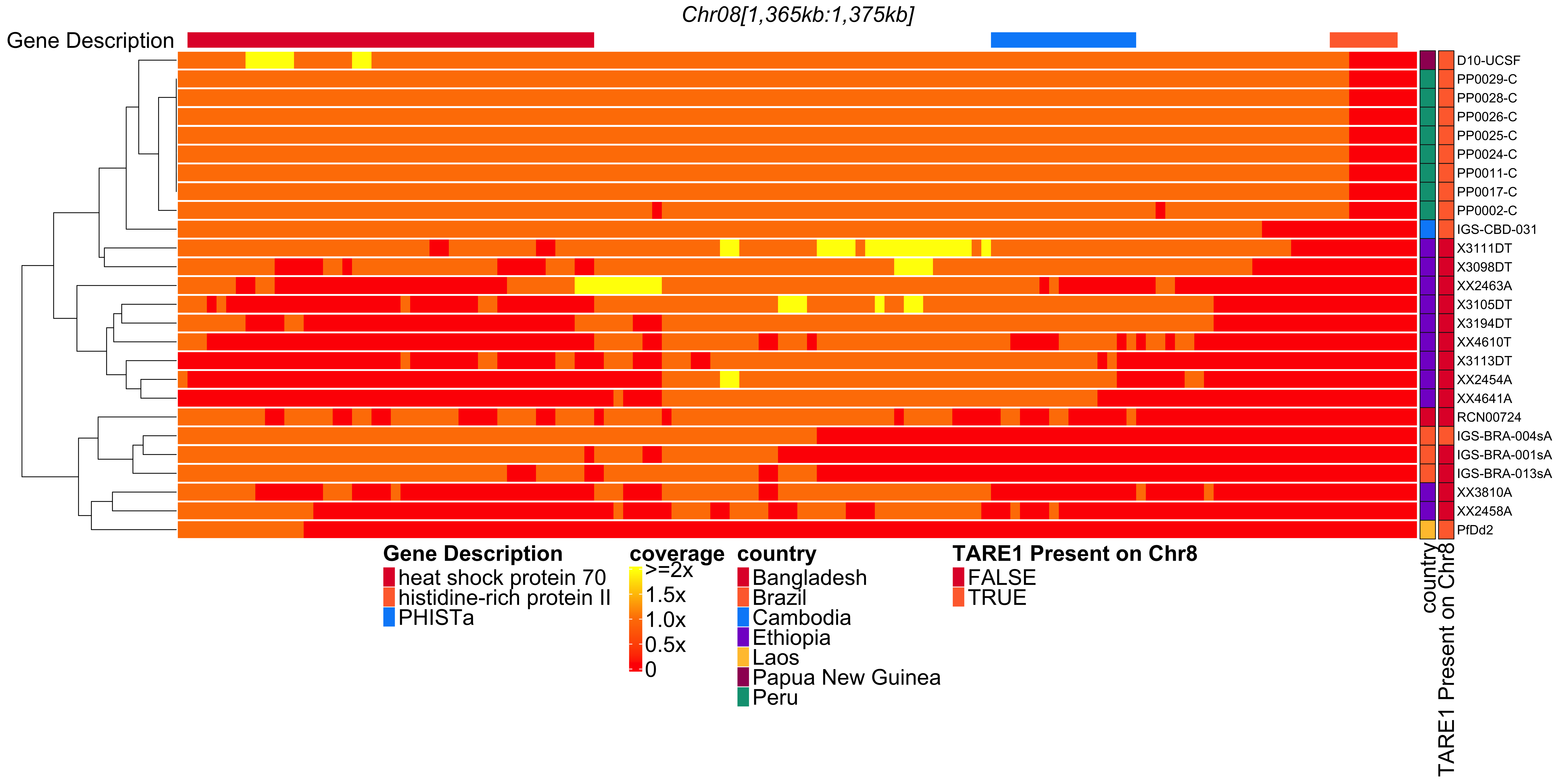
Code
cairo_pdf("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100_cairo.pdf", width = 20, height = 10)
draw(allReports_sp_mat_hm, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", padding = unit(c(25, 2, 2, 2), "mm"))
dev.off()quartz_off_screen
2 Code
pdf("finalHRPII_HRPIII_windows_investTelemeraseHealing_chr8_full_out_step50_windowSize100.pdf", useDingbats = F, width = 20, height = 10)
draw(allReports_sp_mat_hm, background = "transparent", merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom", padding = unit(c(25, 2, 2, 2), "mm"))
dev.off()quartz_off_screen
2 Code
Code
allReports_withTare1 = allReports %>%
filter(sample %in% hrp2Del_samplesWithTare1$sample) %>%
filter(perBaseCoverageNormRounded >0)
allReports_withTare1_filt = allReports_withTare1 %>%
group_by(sample) %>%
slice_max(order_by = start)
allReports_withTare1_filt_sel = allReports_withTare1_filt %>%
select(sample, name)
create_dt(allReports_withTare1_filt_sel)Code
allReports_withTare1_filt_sel_sum = allReports_withTare1_filt_sel %>%
group_by(name) %>%
summarise(n = n())
create_dt(allReports_withTare1_filt_sel_sum)