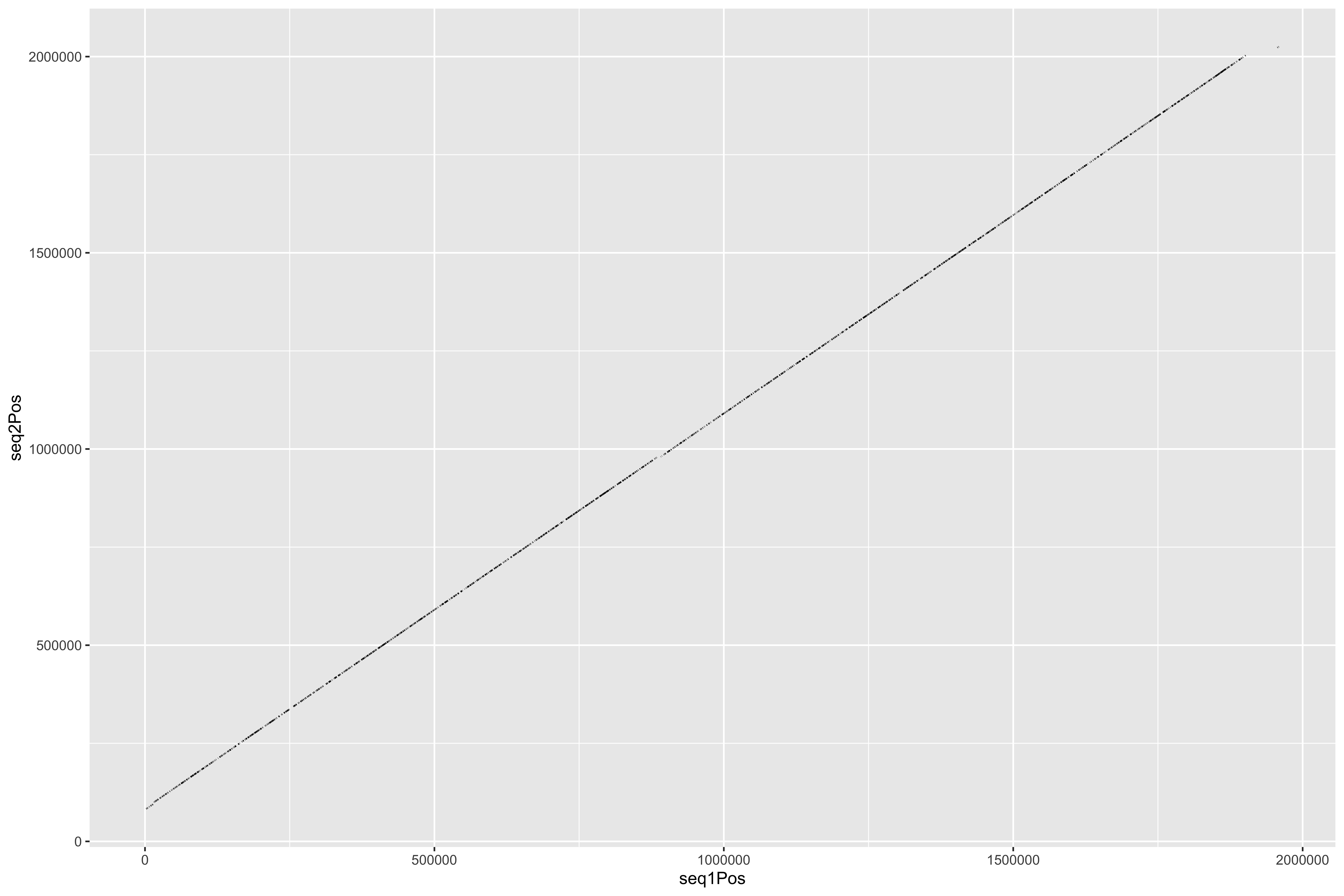
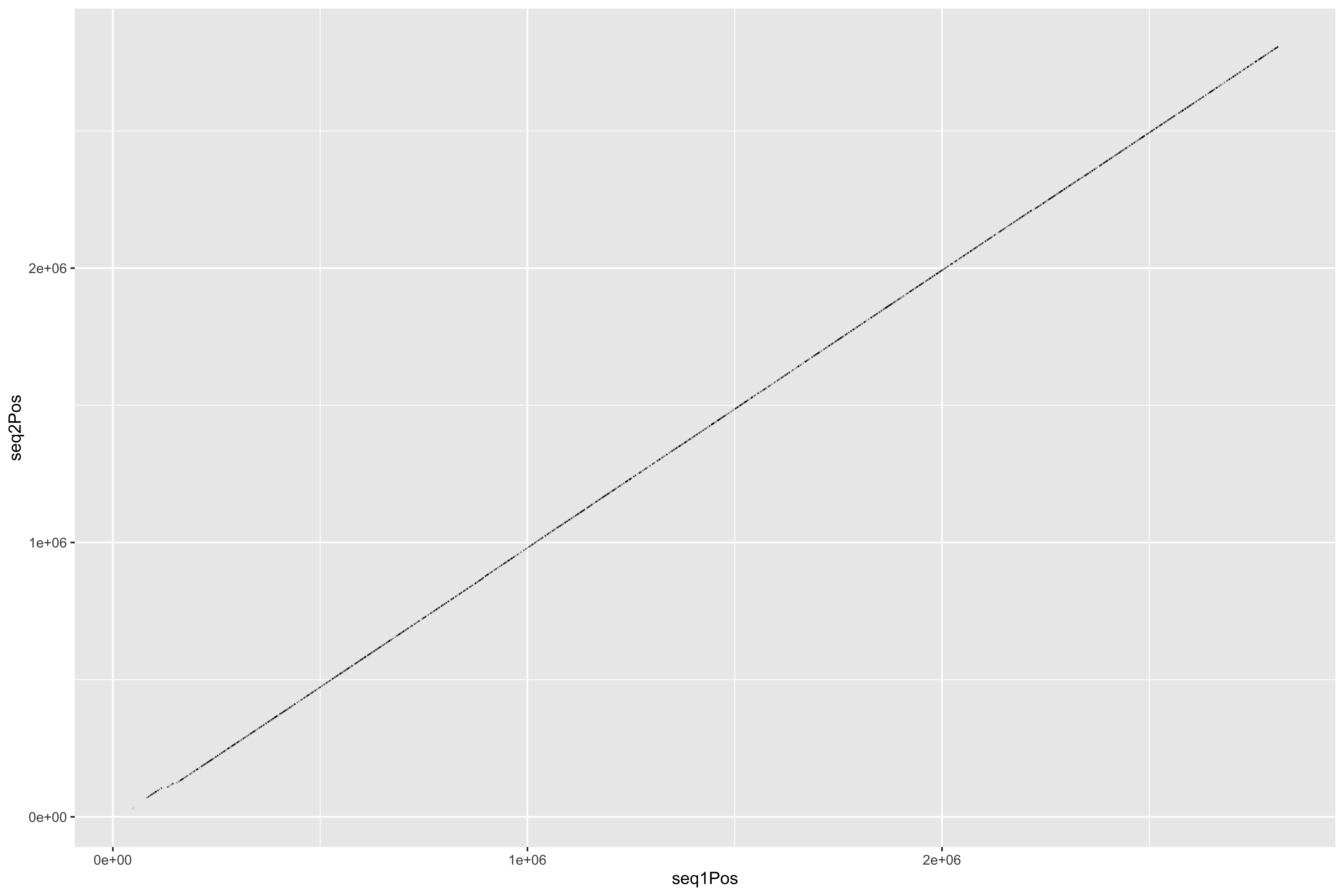
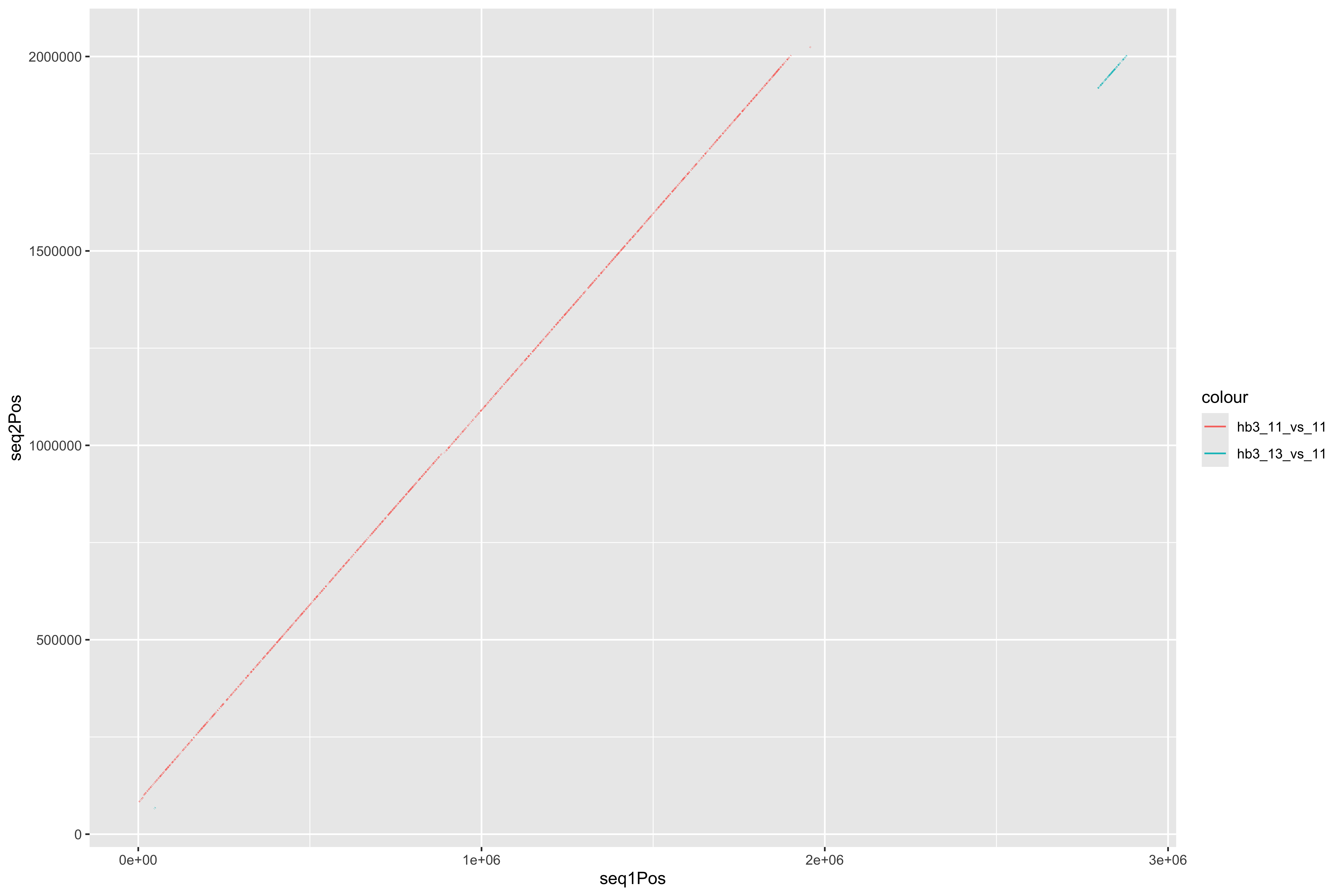
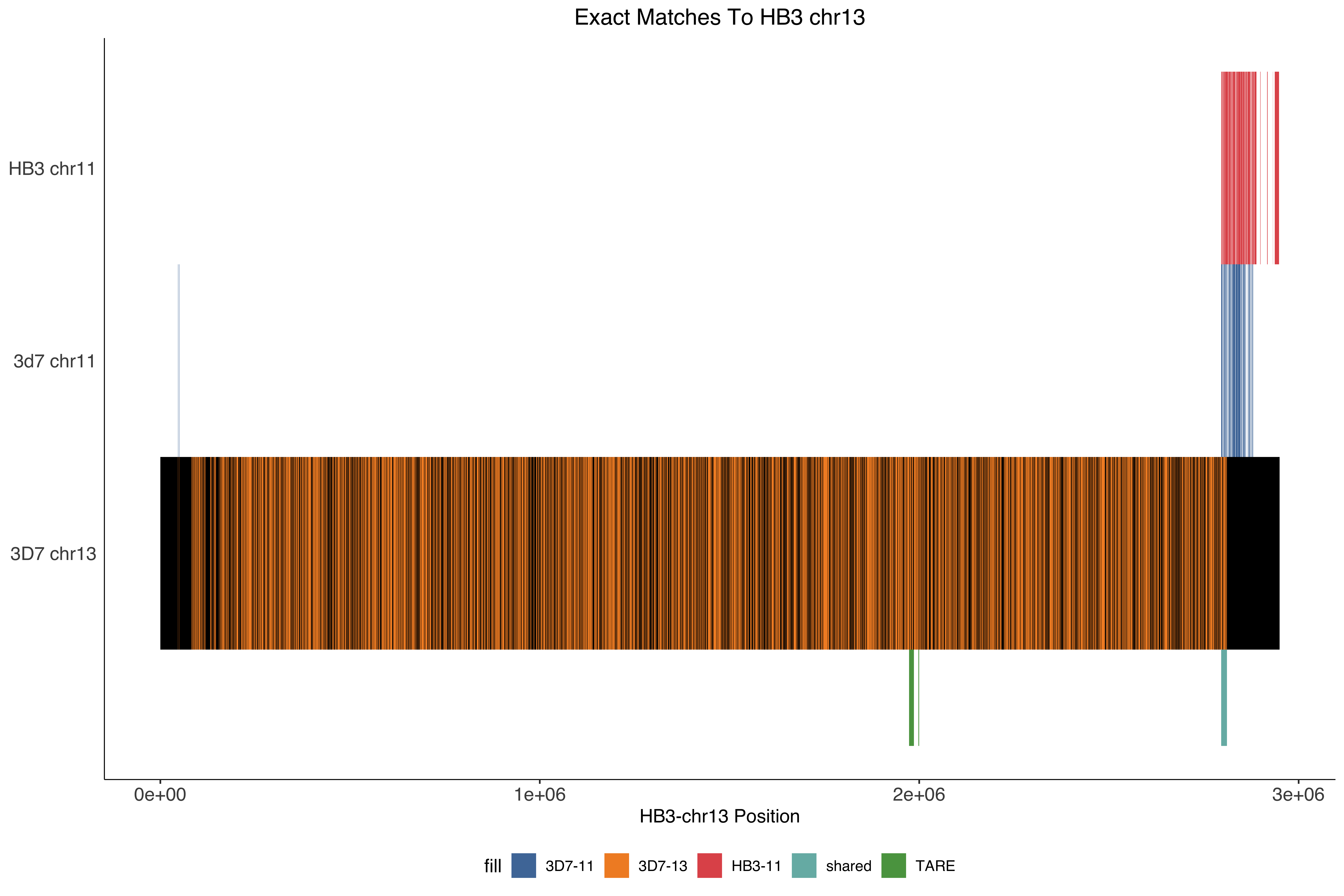
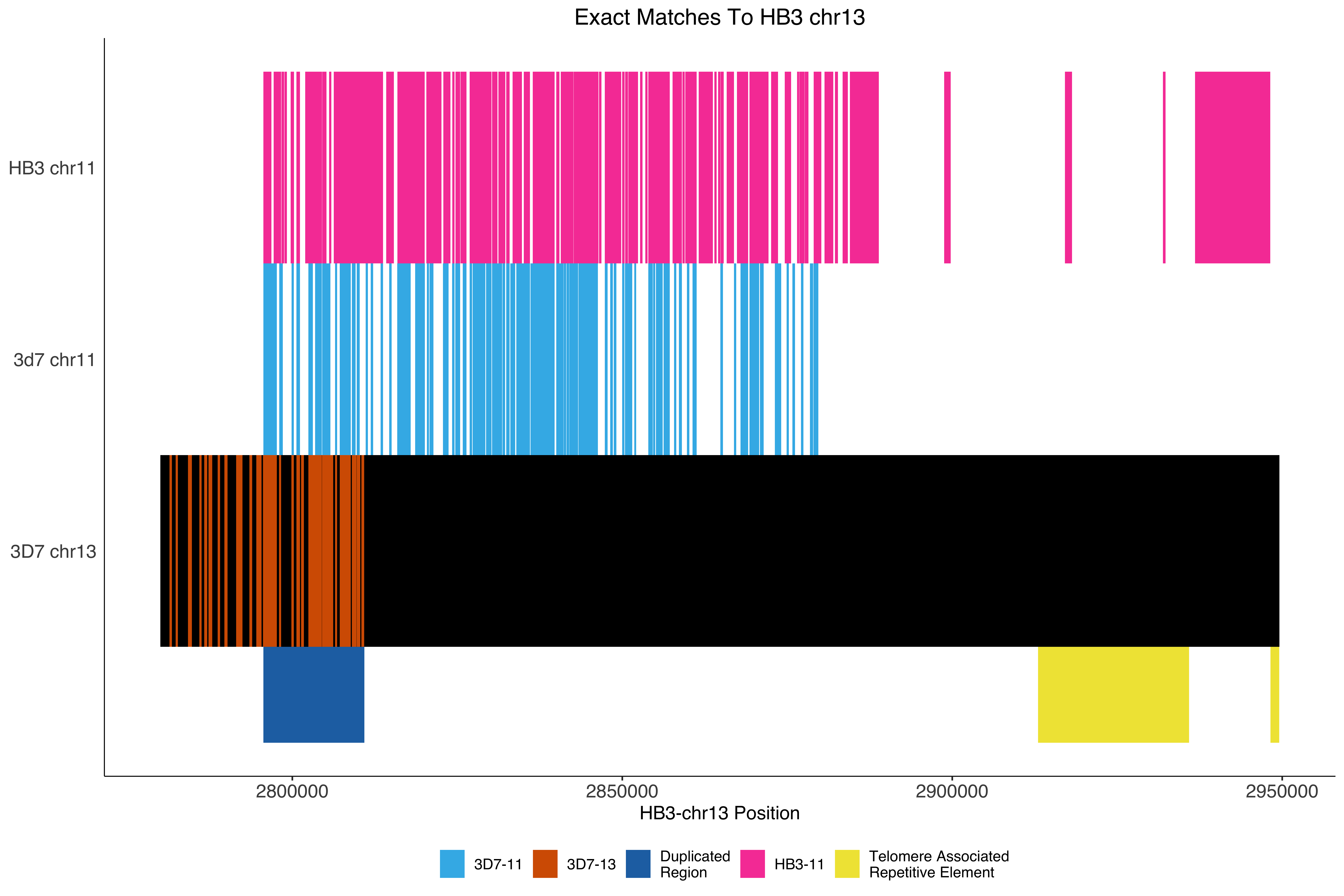
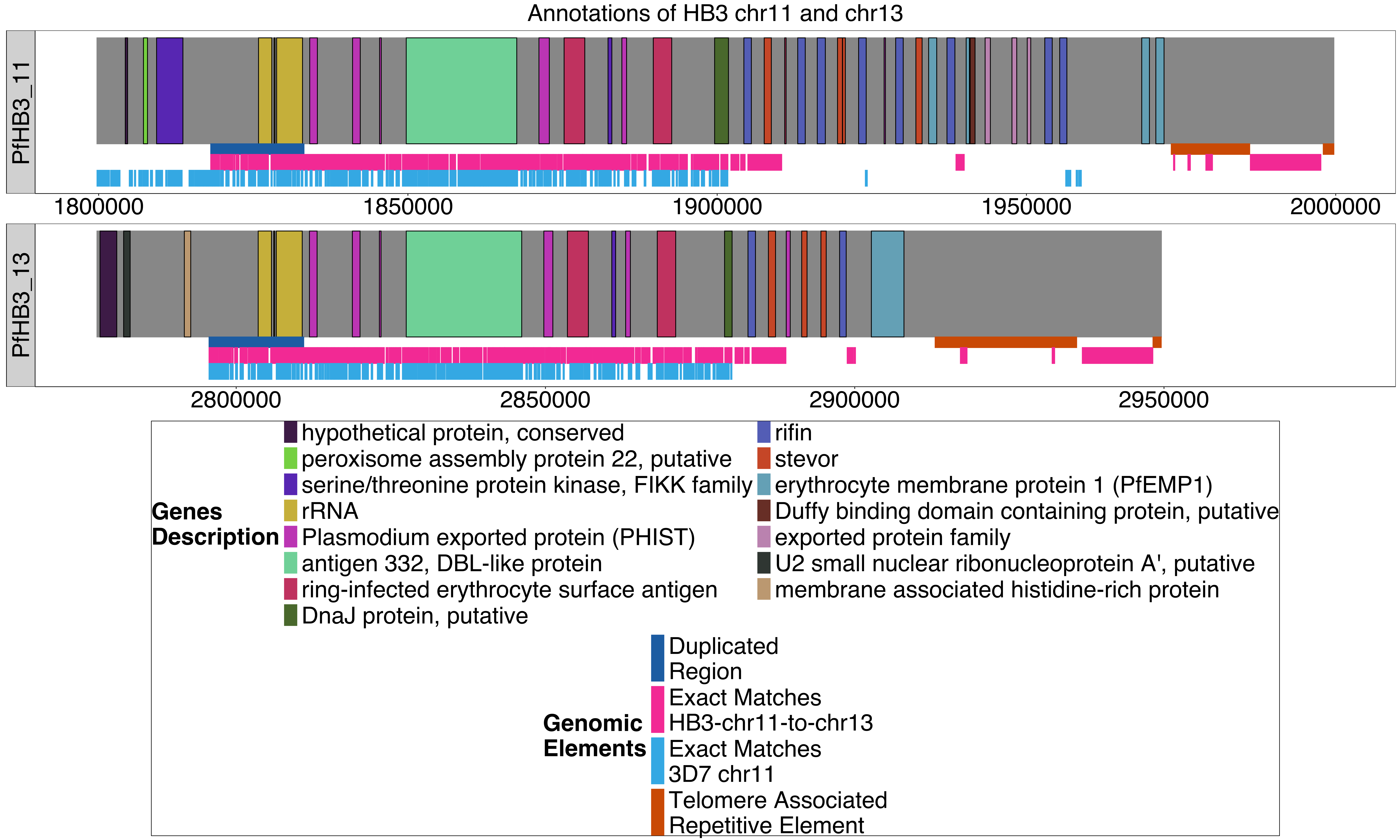
--- title: Processing HB3 Nanopore assembly of chromosome 11 and 13 code-fold: true --- ```{r setup, echo=FALSE, message=FALSE} source("../common.R") ``` ```{bash, eval = F} cat HB3_chr1*.fasta > PfHB3_nanopore_11_13.fasta gsed -i 's/tig00000003/PfHB3_11/g' PfHB3_nanopore_11_13.fasta gsed -i 's/tig00000002/PfHB3_13/g' PfHB3_nanopore_11_13.fasta ``` ```{bash, eval = F} elucidator getKmerDetailedKmerDistAgainstRef --fasta PfHB3_nanopore_11_13.fasta --ref /tank/data/genomes/plasmodium//genomes/pf/genomes/Pf3D7.fasta --trimAtWhiteSpace --getRevComp --kmerLength 19 --overWrite --out dist_agsint_Pf3D7_k19.tab.txt --numThreads 15 elucidator getKmerDetailedKmerDistAgainstRef --fasta PfHB3_nanopore_11_13.fasta --ref /tank/data/genomes/plasmodium//genomes/pf/genomes/PfHB3.fasta --trimAtWhiteSpace --getRevComp --kmerLength 19 --overWrite --out dist_agsint_previousPfHB3_k19.tab.txt --numThreads 15 # reorient chr11 to the 3d7 direction elucidator reOrientReads --fasta PfHB3_nanopore_11_13.fasta --ref /tank/data/genomes/plasmodium//genomes/pf/genomes/Pf3D7.fasta --kLength 11 --overWrite gsed -i 's/_Comp//g' reOriented_PfHB3_nanopore_11_13.fasta nohup elucidator getUniqKmerBlocksOnGenomeAgainstRef --refGenome /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta --trimNameAtWhiteSpace --compGenome reOriented_PfHB3_nanopore_11_13.fasta --kmerLength 19 --numThreads 15 --out compAgainstPf3d7 --overWrite & mkdir fakeGenomes ln -s ../reOriented_PfHB3_nanopore_11_13.fasta . ln -s /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta . nohup elucidator getKmerSharedBlocksBetweenGenomes --genomeDir ./ --primaryGenome Pf3D7 --klen 19 --dout compAgainst3D7 --numThreads 15 & nohup elucidator getKmerSharedBlocksBetweenGenomes --genomeDir ./ --primaryGenome reOriented_PfHB3_nanopore_11_13 --klen 19 --dout compAgainstNewHB3 --numThreads 15 & elucidator extractRefSeqsFromGenomes --bed "/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/hrps/windowSelectionSurroundingHRPs/mappingOutSurroundingRegions/endBeds/Pf3D7_chrom11_toEnd_all.bed" --genomeDir genomesOnlys/ --primaryGenome Pf3D7 --keepBestOnly --shortNames --combineByGenome --numThreads 3 --outputDir extractedChrom11End elucidator extractFromGenomesAndCompare --genomeDir fakeGenomes/ --genomes reOriented_PfHB3_nanopore_11_13 --numThreads 2 --target sharedRegion --sample Pf3D7 --program exact --verbose --dout sharedRegionBlast --fasta ../sharedBetween11_and_13/investigatingChrom11Chrom13/shared_11_13_region.fasta ``` ```{bash, eval = F} elucidator splitFile --trimAtWhiteSpace --fasta Pf3D7.fasta elucidator splitFile --trimAtWhiteSpace --fasta reOriented_PfHB3_nanopore_11_13.fasta elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 PfHB3_11.fasta --seq2 Pf3D7_11_v3.fasta --out 11_vs_11_klen19 --collapse --overWrite elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 PfHB3_11.fasta --seq2 Pf3D7_13_v3.fasta --out 11_vs_13_klen19 --collapse --overWrite elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 PfHB3_13.fasta --seq2 Pf3D7_11_v3.fasta --out 13_vs_11_klen19 --collapse --overWrite elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 PfHB3_13.fasta --seq2 Pf3D7_13_v3.fasta --out 13_vs_13_klen19 --collapse --overWrite elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 PfHB3_11.fasta --seq2 PfHB3_13.fasta --out HB3_11_vs_HB3_13_klen19 --collapse --overWrite elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 19 --seq1 PfHB3_13.fasta --seq2 PfHB3_11.fasta --out HB3_13_vs_HB3_11_klen19 --collapse --overWrite elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 PfHB3_11.fasta --seq2 Pf3D7_11_v3.fasta --out 11_vs_11_klen300 --collapse --overWrite elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 PfHB3_11.fasta --seq2 Pf3D7_13_v3.fasta --out 11_vs_13_klen300 --collapse --overWrite elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 PfHB3_13.fasta --seq2 Pf3D7_11_v3.fasta --out 13_vs_11_klen300 --collapse --overWrite elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 PfHB3_13.fasta --seq2 Pf3D7_13_v3.fasta --out 13_vs_13_klen300 --collapse --overWrite elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 PfHB3_11.fasta --seq2 PfHB3_13.fasta --out HB3_11_vs_HB3_13_klen300 --collapse --overWrite elucidator findPositionsOfUniqueKmersInEachOther --kmerLength 300 --seq1 PfHB3_13.fasta --seq2 PfHB3_11.fasta --out HB3_13_vs_HB3_11_klen300 --collapse --overWrite mummer -b -c -F -l 200 -maxmatch PfHB3_11.fasta Pf3D7_11_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfHB3_11_vs_Pf3D7_11_exactMatches_minlen200.tab.txt mummer -b -c -F -l 200 -maxmatch PfHB3_11.fasta Pf3D7_13_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfHB3_11_vs_Pf3D7_13_exactMatches_minlen200.tab.txt mummer -b -c -F -l 200 -maxmatch PfHB3_13.fasta Pf3D7_11_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfHB3_13_vs_Pf3D7_11_exactMatches_minlen200.tab.txt mummer -b -c -F -l 200 -maxmatch PfHB3_13.fasta Pf3D7_13_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfHB3_13_vs_Pf3D7_13_exactMatches_minlen200.tab.txt mummer -b -c -F -l 200 -maxmatch PfHB3_11.fasta PfHB3_13.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > HB3_11_vs_HB3_13_exactMatches_minlen200.tab.txt mummer -b -c -F -l 200 -maxmatch PfHB3_13.fasta PfHB3_11.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > HB3_13_vs_HB3_11_exactMatches_minlen200.tab.txt mummer -b -c -F -l 200 PfHB3_11.fasta Pf3D7_11_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfHB3_11_vs_Pf3D7_11_exactUniqueMatches_minlen200.tab.txt mummer -b -c -F -l 200 PfHB3_11.fasta Pf3D7_13_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfHB3_11_vs_Pf3D7_13_exactUniqueMatches_minlen200.tab.txt mummer -b -c -F -l 200 PfHB3_13.fasta Pf3D7_11_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfHB3_13_vs_Pf3D7_11_exactUniqueMatches_minlen200.tab.txt mummer -b -c -F -l 200 PfHB3_13.fasta Pf3D7_13_v3.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > PfHB3_13_vs_Pf3D7_13_exactUniqueMatches_minlen200.tab.txt mummer -b -c -F -l 200 PfHB3_11.fasta PfHB3_13.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > HB3_11_vs_HB3_13_exactUniqueMatches_minlen200.tab.txt mummer -b -c -F -l 200 PfHB3_13.fasta PfHB3_11.fasta | elucidator parseMummberResultsToBed --mummerOut STDIN | elucidator splitColumnContainingMeta --file STDIN --delim tab --removeEmptyColumn --header --column meta > HB3_13_vs_HB3_11_exactUniqueMatches_minlen200.tab.txt elucidator getReadLens --fasta Pf3D7.fasta --trimAtWhiteSpace > chroms_Pf3D7.txt elucidator getReadLens --fasta reOriented_PfHB3_nanopore_11_13.fasta --trimAtWhiteSpace > chroms_reOriented_PfHB3_nanopore_11_13.txt ``` ```{r} = 50000 = backtraceAmount= readr:: read_tsv ("../rRNA_segmental_duplications/sharedBetween11_and_13/investigatingChrom11Chrom13/shared_11_13_region.bed" , col_names = T)= readr:: read_tsv ("fakeGenomes/HB3_11_vs_HB3_13_exactUniqueMatches_minlen200.tab.txt" ) %>% filter (strand == "+" )= readr:: read_tsv ("fakeGenomes/HB3_13_vs_HB3_11_exactUniqueMatches_minlen200.tab.txt" ) %>% filter (strand == "+" )= readr:: read_tsv ("fakeGenomes/PfHB3_11_vs_Pf3D7_11_exactUniqueMatches_minlen200.tab.txt" ) %>% left_join (sharedRegion_3d7 %>% rename (query = ` #chrom ` , sharedStart = start, sharedEnd = end) %>% select (query, sharedStart, sharedEnd)) %>% filter (queryStart >= sharedStart - backtrace)= readr:: read_tsv ("fakeGenomes/PfHB3_13_vs_Pf3D7_11_exactUniqueMatches_minlen200.tab.txt" ) %>% filter (strand == "+" ) %>% left_join (sharedRegion_3d7 %>% rename (query = ` #chrom ` , sharedStart = start, sharedEnd = end) %>% select (query, sharedStart, sharedEnd)) %>% filter (queryStart >= sharedStart - backtrace)= readr:: read_tsv ("fakeGenomes/chroms_Pf3D7.txt" , col_names = c ( "chrom" , "len" ))= readr:: read_tsv ("fakeGenomes/chroms_reOriented_PfHB3_nanopore_11_13.txt" , col_names = c ( "chrom" , "len" ))= readr:: read_tsv ("fakeGenomes/11_vs_11_klen300.tab.txt" )ggplot (hb3_11_vs_11) + geom_segment (aes (x = seq1Pos, xend = seq1Pos + size, y = seq2Pos, yend = seq2Pos + size))= readr:: read_tsv ("fakeGenomes/13_vs_13_klen300.tab.txt" )ggplot (hb3_13_vs_13) + geom_segment (aes (x = seq1Pos, xend = seq1Pos + size, y = seq2Pos, yend = seq2Pos + size))= readr:: read_tsv ("fakeGenomes/11_vs_13_klen300.tab.txt" )ggplot (hb3_11_vs_13) + geom_segment (aes (x = seq1Pos, xend = seq1Pos + size, y = seq2Pos, yend = seq2Pos + size))= readr:: read_tsv ("fakeGenomes/13_vs_11_klen300.tab.txt" )ggplot (hb3_13_vs_11) + geom_segment (aes (x = seq1Pos, xend = seq1Pos + size, y = seq2Pos, yend = seq2Pos + size, color = "hb3_13_vs_11" ))+ geom_segment (aes (x = seq1Pos, xend = seq1Pos + size, y = seq2Pos, yend = seq2Pos + size, color = "hb3_11_vs_11" ), data = hb3_11_vs_11)= readr:: read_tsv ("fakeGenomes/HB3_11_vs_HB3_13_klen300.tab.txt" )= readr:: read_tsv ("fakeGenomes/HB3_13_vs_HB3_11_klen300.tab.txt" )= readr:: read_tsv ("sharedRegionBlast/reOriented_PfHB3_nanopore_11_13/regions.bed" , col_names = F) %>% unique ()= readr:: read_tsv ("sharedRegionBlast/reOriented_PfHB3_nanopore_11_13/regions.bed" , col_names = F) %>% unique () %>% filter (X1 == "PfHB3_13" )= readr:: read_tsv ("sharedRegionBlast/reOriented_PfHB3_nanopore_11_13/regions.bed" , col_names = F) %>% unique () %>% filter (X1 == "PfHB3_11" )= readr:: read_tsv ("repeats_filt_starts_ends_tares.tsv" ) %>% mutate (X1 = gsub ("PfHB3nano" , "PfHB3" , X1))= tares %>% filter (X1 == "PfHB3_11" ) %>% filter (! is.na (endPos))= tares %>% filter (X1 == "PfHB3_13" ) %>% filter (! is.na (endPos))= bind_rows (ggplot () + geom_rect (aes (xmin = 0 , xmax = len, ymin = 0 , ymax = 1 ), fill = "grey0" ,data = HB3chromsLens %>% filter (chrom == "PfHB3_13" )) + geom_rect (aes (xmin = seq1Pos, xmax = seq1Pos + size, ymin = 0 , ymax = 1 , fill = "3D7-13" ), data = hb3_13_vs_13) + geom_rect (aes (xmin = seq1Pos, xmax = seq1Pos + size, ymin = 1 , ymax = 2 , fill = "3D7-11" ), data = hb3_13_vs_11) + geom_rect (aes (xmin = seq1Pos, xmax = seq1Pos + size, ymin = 2 , ymax = 3 , fill = "HB3-11" ), data = hb3_13_vs_hb3_11) + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 0.5 , ymax = 0 , fill = "shared" data = sharedRegion_HB3_13) + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 0.5 , ymax = 0 , fill = "TARE" data = tares_chrom11_end) + scale_fill_tableau () + + theme (#axis.text.y = element_blank(), axis.ticks.y = element_blank (), panel.border = element_blank ()) + scale_y_continuous (breaks = c (0.5 ,1.5 , 2.5 ), labels = c ("3D7 chr13" , "3d7 chr11" , "HB3 chr11" )) + labs (x = "HB3-chr13 Position" , title = "Exact Matches To HB3 chr13" ) = ggplot () + geom_rect (aes (xmin = 2780000 , xmax = len, ymin = 0 , ymax = 1 ), fill = "grey0" ,data = HB3chromsLens %>% filter (chrom == "PfHB3_13" )) + geom_rect (aes (xmin = seq1Pos, xmax = seq1Pos + size, ymin = 0 , ymax = 1 , fill = "3D7-13" ), data = hb3_13_vs_13) + geom_rect (aes (xmin = seq1Pos, xmax = seq1Pos + size, ymin = 1 , ymax = 2 , fill = "3D7-11" ), data = hb3_13_vs_11) + geom_rect (aes (xmin = seq1Pos, xmax = seq1Pos + size, ymin = 2 , ymax = 3 , fill = "HB3-11" ), data = hb3_13_vs_hb3_11) + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 0.5 , ymax = 0 , fill = "Duplicated \n Region" data = sharedRegion_HB3_13) + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 0.5 , ymax = 0 , fill = "Telomere Associated \n Repetitive Element" data = tares_chrom13_end) + #scale_fill_tableau() + scale_fill_manual (values = c ("3D7-11" = "#3DB7E9" , "3D7-13" = "#D55E00" , "HB3-11" = "#F748A5" , "Duplicated \n Region" = "#2271B2" , "Telomere Associated \n Repetitive Element" = "#F0E442" )) + + theme (#axis.text.y = element_blank(), axis.ticks.y = element_blank (), panel.border = element_blank ()) + scale_y_continuous (breaks = c (0.5 ,1.5 , 2.5 ), labels = c ("3D7 chr13" , "3d7 chr11" , "HB3 chr11" )) + labs (x = "HB3-chr13 Position" ,title = "Exact Matches To HB3 chr13" , fill = "" ) + xlim (2780000 ,2949590 ) pdf ("zoomInHB3_chr13_exactMatchesPlot.pdf" , width = 11 , height = 8 , useDingbats = F)print (zoomInHB3_chr13_exactMatchesPlot)dev.off ()= ggplot () + geom_rect (aes (xmin = 1810000 , xmax = len, ymin = 0 , ymax = 1 ), fill = "grey0" ,data = HB3chromsLens %>% filter (chrom == "PfHB3_11" )) + geom_rect (aes (xmin = seq1Pos, xmax = seq1Pos + size, ymin = 1 , ymax = 2 , fill = "3D7-13" ), data = hb3_11_vs_13) + geom_rect (aes (xmin = seq1Pos, xmax = seq1Pos + size, ymin = 0 , ymax = 1 , fill = "3D7-11" ), data = hb3_11_vs_11) + geom_rect (aes (xmin = seq1Pos, xmax = seq1Pos + size, ymin = 2 , ymax = 3 , fill = "HB3-13" ), data = hb3_11_vs_hb3_13) + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 0.5 , ymax = 0 , fill = "Duplicated \n Region" data = sharedRegion_HB3_11) + scale_fill_tableau () + + theme (#axis.text.y = element_blank(), axis.ticks.y = element_blank (), panel.border = element_blank ()) + scale_y_continuous (breaks = c (0.5 ,1.5 , 2.5 ), labels = c ("3D7 chr11" , "3d7 chr13" , "HB3 chr13" )) + labs (x = "HB3-chr11 Position" , title = "Exact Matches To HB3 chr11" ) + xlim (1810000 ,1999710 ) ``` ```{bash, eval = F} cd sharedRegionBlast/reOriented_PfHB3_nanopore_11_13 elucidator getReadLens --fasta ../../reOriented_PfHB3_nanopore_11_13.fasta > chromLens.txt elucidator extendToEndOfChrom --bed uniq_regions.bed --chromLengthsTable chromLens.txt --out uniq_regions_toEnd.bed elucidator extendBedRegions --bed uniq_regions_toEnd.bed --left 50000 --out l50000_uniq_regions_toEnd.bed elucidator bedGetIntersectingRecordsInGff --bed l50000_uniq_regions_toEnd.bed --gff ../../annotations/scaffold.out.gff3 --extraAttributes product --overWrite --features polypeptide,rRNA --out gene_out elucidator expandTableBySeparatingColumn --fnp gene_out.bed --sep "],[" --columnName col.6 --delim tab | sed 's/.*ID=//g' | sed 's/;.*//g' | sort | uniq > ids_near_ends.txt elucidator gffToBedByID --id ids_near_ends.txt --gff ../../annotations/scaffold.out.gff3 --overWrite --out ids_near_ends.bed elucidator bedGetIntersectingRecordsInGff --bed ids_near_ends.bed --gff ../../annotations/scaffold.out.gff3 --extraAttributes product --overWrite --features polypeptide,rRNA --out ids_near_ends_withInfo sed 's/evidence.*;//g' ids_near_ends_withInfo.bed > renamed_ids_near_ends_withInfo.bed elucidator splitColumnContainingMeta --removeEmptyColumn --delim tab --column col.6 --file renamed_ids_near_ends_withInfo.bed --addHeader --out renamed_ids_near_ends_withInfo.tab.txt --overWrite ``` ```{bash, eval = F} elucidator extractRefSeqsFromGenomes --bed "/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/rRNA/all/merged_updatedrRNA_locs.bed" --genomeDir fakeGenomes/ --primaryGenome Pf3D7 --outputDir extract_rRNA_regions --numThreads 15 --overWriteDirs --selectedGenomes Pf3D7,reOriented_PfHB3_nanopore_11_13 elucidator extractRefSeqsFromGenomes --bed "/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/rRNA/all/28s_rRNA_locs.bed" --genomeDir fakeGenomes/ --primaryGenome Pf3D7 --outputDir extract_28s_rRNA_regions --numThreads 15 --overWriteDirs --selectedGenomes Pf3D7,reOriented_PfHB3_nanopore_11_13 ``` # Gene Annotations ```{r} #| code-fold: true = readr:: read_tsv ("extract_28s_rRNA_regions/Pf3D7_11_v3-1928933-1933138-for/beds/reOriented_PfHB3_nanopore_11_13_region.bed" , col_names = F) %>% rename (col.0 = X1, col.1 = X2, col.2 = X3, col.3 = X4, col.4 = X5, col.5 = X6) %>% mutate (ID = "PfHB3_rRNA-28s" , feature = "rRNA" , product = "rRNA" , product_mod = "rRNA" )= readr:: read_tsv ("sharedRegionBlast/reOriented_PfHB3_nanopore_11_13/renamed_ids_near_ends_withInfo.tab.txt" )= annotations %>% mutate (product_mod = gsub ("term=" , "" , product)) %>% #rename(chrom = col.0, start = col.1, end = col.2, name = col.3, length = col.4, strand = col.5) %>% # mutate(gene = ifelse(grepl("PF3D7_0831800", product_mod), "HRP II", "other")) %>% # mutate(gene = ifelse(grepl("PF3D7_1372200", product_mod), "HRP III", gene)) %>% mutate (product_mod = gsub ("unknownfunction" , "unknown function" , product_mod)) %>% mutate (product_mod = gsub (" \\ (SURFIN" , " \\ (SURFIN" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("Plasmodium exported protein" , product_mod), "Plasmodium exported protein (PHIST)" , product_mod)) %>% mutate (product_mod = ifelse ("erythrocyte membrane protein 1 (PfEMP1), exon 2" == product_mod, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene" , product_mod)) %>% mutate (product_mod = ifelse ("membrane associated erythrocyte binding-like protein" == product_mod, "merozoite adhesive erythrocytic binding protein" , product_mod)) %>% mutate (product_mod = ifelse ("membrane associated erythrocyte binding-likeprotein" == product_mod, "merozoite adhesive erythrocytic binding protein" , product_mod)) %>% mutate (product_mod = ifelse ("membrane associated histidine-rich protein 1" == product_mod, "membrane associated histidine-rich protein" , product_mod)) %>% mutate (product_mod = gsub (",putative" , ", putative" , product_mod)) %>% mutate (product_mod = gsub (",pseudogene" , ", pseudogene" , product_mod))%>% mutate (product_mod = gsub ("unknownfunction" , "unknown function" , product_mod))%>% mutate (product_mod = gsub ("conserved protein, unknown function" , "conserved Plasmodium protein, unknown function" , product_mod)) %>% mutate (product_mod = gsub (" \\ (SURFIN" , " \\ (SURFIN" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("Plasmodium exported protein" , product_mod), "Plasmodium exported protein (PHIST)" , product_mod)) %>% mutate (product_mod = ifelse ("erythrocyte membrane protein 1 (PfEMP1), exon 2" == product_mod, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene" , product_mod)) %>% mutate (product_mod = ifelse ("membrane associated histidine-rich protein 1" == product_mod, "membrane associated histidine-rich protein" , product_mod))%>% mutate (product_mod = gsub (",pseudogene" , ", pseudogene" , product_mod))%>% mutate (product_mod = gsub ("unknownfunction" , "unknown function" , product_mod))%>% mutate (product_mod = gsub (" \\ (SURFIN" , " \\ (SURFIN" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("Plasmodium exported protein" , product_mod), "Plasmodium exported protein (PHIST)" , product_mod)) %>% mutate (product_mod = ifelse ("erythrocyte membrane protein 1 (PfEMP1), exon 2" == product_mod, "erythrocyte membrane protein 1 (PfEMP1), exon 2, pseudogene" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("sporozoite and liver stage tryptophan-rich protein, putative" , product_mod), "tryptophan/threonine-rich antigen" , product_mod))%>% mutate (product_mod = ifelse (grepl ("CRA domain-containing protein, putative" , product_mod), "conserved Plasmodium protein, unknown function" , product_mod))%>% mutate (product_mod = gsub (",pseudogene" , ", pseudogene" , product_mod)) %>% mutate (product_mod = gsub ("surfaceantigen" , "surface antigen" , product_mod)) %>% mutate (product_mod = gsub ("Tetratricopeptide repeat, putative" , "tetratricopeptide repeat protein, putative" , product_mod)) %>% mutate (product_mod = gsub ("transmembraneprotein" , "transmembrane protein" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("PfEMP1" , product_mod) & grepl ("pseudogene" , product_mod), "erythrocyte membrane protein 1 (PfEMP1), pseudogene" , product_mod)) %>% mutate (product_mod = gsub ("PIR protein" , "stevor" , product_mod)) %>% mutate (product_mod = gsub ("erythrocyte membrane protein 1-like" , "erythrocyte membrane protein 1 (PfEMP1), pseudogene" , product_mod)) %>% mutate (product_mod = gsub ("acidic terminal segments, variant surface antigen of PfEMP1, putative" , "erythrocyte membrane protein 1 (PfEMP1), pseudogene" , product_mod))%>% mutate (product_mod = ifelse (grepl ("CoA binding protein" , product_mod, ignore.case = T), "acyl-CoA binding protein" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("transfer RNA" , product_mod) | grepl ("tRNA" , product_mod), "tRNA" , product_mod))%>% mutate (product_mod = ifelse (grepl ("cytoadherence" , product_mod), "CLAG" , product_mod))%>% mutate (product_mod = ifelse (grepl ("surface-associated interspersed protein" , product_mod), "SURFIN" , product_mod))%>% mutate (product_mod = ifelse (grepl ("SURFIN" , product_mod), "SURFIN" , product_mod))%>% mutate (product_mod = ifelse (grepl ("stevor-like" , product_mod), "stevor, pseudogene" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("exported protein family" , product_mod), "exported protein family" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("rRNA" , feature), "rRNA" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("serine/threonine protein kinase" , product_mod), "serine/threonine protein kinase, FIKK family" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("hypothetical protein" , product_mod), "hypothetical protein, conserved" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("conserved Plasmodium protein, unknown function" , product_mod), "hypothetical protein, conserved" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("Rifin/stevor family, putative" , product_mod), "stevor" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("erythrocyte membrane protein 1 (PfEMP1), pseudogene" , product_mod), "erythrocyte membrane protein 1 (PfEMP1)" , product_mod)) %>% mutate (product_mod = ifelse (grepl ("erythrocyte membrane protein 1" , product_mod), "erythrocyte membrane protein 1 (PfEMP1)" , product_mod))%>% mutate (product_mod = ifelse (grepl ("Plasmodium RESA N-terminal, putative" , product_mod), "ring-infected erythrocyte surface antigen" , product_mod)) %>% left_join (rRNA_28s %>% select (col.0 , col.1 , col.2 ) %>% rename (` 28rRNA-start ` = col.1 , ` 28rRNA-end ` = col.2 )) %>% filter (! (col.1 >= ` 28rRNA-start ` & col.1 <= ` 28rRNA-end ` )) %>% bind_rows (rRNA_28s)= annotations %>% filter (ID %!in% c ("PfHB3_110054900.1:pep" , "PfHB3_110055000.1:pep" )) %>% filter ((col.0 == "PfHB3_11" & col.1 >= 1849728 - backtraceAmount) | .0 == "PfHB3_13" & col.1 >= 2827490 - backtraceAmount)) %>% filter (! ("hypothetical protein, conserved" == product_mod & .0 == "PfHB3_11" & col.1 >= 1944764 ) | .0 == "PfHB3_13" & col.1 >= 2859471 ))))= tibble (col.0 = c ("PfHB3_11" , "PfHB3_13" ), col.1 = c (1849728 - backtraceAmount, 2827490 - backtraceAmount))= HB3chromsLens %>% rename (col.0 = chrom, col.2 = len) %>% left_join (annotations_filt_blank %>% select (col.0 , col.1 ) %>% rename (start = col.1 )) %>% mutate (len = col.2 - start)= annotations_filt_blank %>% left_join (HB3chromsLens_mod %>% select (col.0 , len)) %>% mutate (col.2 = col.1 + max (len)) %>% mutate (newLen = col.2 - col.1 )= bind_rows (%>% select (seq1, seq1Pos, size) %>% rename (col.0 = seq1, col.1 = seq1Pos) %>% mutate (col.2 = col.1 + size), %>% select (seq2, seq2Pos, size) %>% rename (col.0 = seq2, col.1 = seq2Pos) %>% mutate (col.2 = col.1 + size)= bind_rows (%>% select (seq1, seq1Pos, size) %>% rename (col.0 = seq1, col.1 = seq1Pos) %>% mutate (col.2 = col.1 + size) %>% filter (col.1 >= 1849728 - backtraceAmount), %>% select (seq1, seq1Pos, size) %>% rename (col.0 = seq1, col.1 = seq1Pos) %>% mutate (col.2 = col.1 + size) %>% filter (col.1 >= 2827490 - backtraceAmount)= bind_rows (%>% select (` #target ` , targetStart, length, query, queryStart, queryEnd) %>% rename (col.0 = ` #target ` , col.1 = targetStart, other = query, otherStart = queryStart, otherEnd = queryEnd%>% mutate (col.2 = col.1 + length), %>% select (query, queryStart, length, ` #target ` , targetStart, targetEnd) %>% rename (col.0 = query, col.1 = queryStart, other = ` #target ` , otherStart = targetStart, otherEnd = targetEnd%>% mutate (col.2 = col.1 + length)%>% mutate (start = col.1 ,end = col.2 )= bind_rows (%>% select (` #target ` , targetStart, length, query, queryStart, queryEnd) %>% rename (col.0 = ` #target ` , col.1 = targetStart, other = query, otherStart = queryStart, otherEnd = queryEnd%>% mutate (col.2 = col.1 + length)%>% filter (col.1 >= 1849728 - backtraceAmount), %>% select (` #target ` , targetStart, length, query, queryStart, queryEnd) %>% rename (col.0 = ` #target ` , col.1 = targetStart, other = query, otherStart = queryStart, otherEnd = queryEnd) %>% mutate (col.2 = col.1 + length)%>% filter (col.1 >= 2827490 - backtraceAmount)%>% mutate (start = col.1 ,end = col.2 )= scheme$ hex (length (unique (c (annotations_filt$ product_mod))))names (rawGeneAnnotationsColors) = unique (c (annotations_filt$ product_mod))write_tsv (tibble (names = names (rawGeneAnnotationsColors), colors = rawGeneAnnotationsColors), "rawGeneAnnotationsColors.tab.txt" )= c ()"Duplicated \n Region" ] = "#2271B2" "Exact Matches \n HB3-chr11-to-chr13" ] = "#F748A5" "Exact Matches \n 3D7 chr11" ] = "#3DB7E9" "Telomere Associated \n Repetitive Element" ] = "#D55E00" # genomicElementsColors["Duplicated\nRegion"] = "#4E79A7" # genomicElementsColors["Exact Matches\nHB3-chr11-to-chr13"] = "#E15759" # genomicElementsColors["Exact Matches\n3D7 chr11"] = "#59A14F" # genomicElementsColors["Telomere Associated\nRepetitive Element"] = "#B07AA1" = c (rawGeneAnnotationsColors, genomicElementsColors)= sharedRegion %>% mutate (start = X2,end = X3)= tares_chrom11_chrom13_ends %>% mutate (start = X2,end = X3)= hb3_13_vs_hb3_11_forPlotting %>% mutate (start = col.1 ,end = col.2 )= hb3_13_and_hb3_11_vs3d7_forPlotting %>% mutate (start = col.1 ,end = col.2 )``` ```{r} = ggplot (annotations_filt) + geom_rect (aes (xmin = col.1 ,xmax = col.2 ,ymin = 0 ,ymax = 1 data = annotations_filt_blank,alpha = 0 ) + geom_rect (aes (xmin = start,xmax = col.2 ,ymin = 0 ,ymax = 1 fill = "grey60" ,color = "grey60" ,data = HB3chromsLens_mod+ geom_rect (aes (xmin = col.1 ,xmax = col.2 ,ymin = 0 ,ymax = 1 ,fill = product_mod,# color = product_mod color = "black" ) + facet_wrap ( ~ col.0 ,scales = "free" ,ncol = 1 ,strip.position = "left" ) + geom_rect (aes (xmin = X2,xmax = X3,ymin = - 0.1 ,ymax = 0 ,start = start,end = end,color = "Duplicated \n Region" ,fill = "Duplicated \n Region" data = sharedRegion %>% mutate (col.0 = X1)+ geom_rect (aes (xmin = X2,xmax = X3,ymin = - 0.1 ,ymax = 0 ,start = start,end = end,color = "Telomere Associated \n Repetitive Element" ,fill = "Telomere Associated \n Repetitive Element" data = tares_chrom11_chrom13_ends %>% mutate (col.0 = X1)+ # geom_rect( # aes( # xmin = col.1, # xmax = col.2, # ymin = -0.25, # ymax = -0.1, # start = start, # end = end, # fill = "Exact Matches\nHB3-chr11-to-chr13" # ), # data = hb3_13_vs_hb3_11_forPlotting # ) + geom_rect (aes (xmin = col.1 ,xmax = col.2 ,ymin = - 0.25 ,ymax = - 0.1 ,start = start,end = end,length = length, other = other, otherStart = otherStart,otherEnd = otherEnd, color = "Exact Matches \n HB3-chr11-to-chr13" ,fill = "Exact Matches \n HB3-chr11-to-chr13" data = hb3_13_vs_hb3_11_exact_forPlotting+ # geom_rect( # aes( # xmin = col.1, # xmax = col.2, # ymin = -0.40, # ymax = -0.25, # start = start, # end = end, # fill = "Exact Matches\n3D7 chr11" # ), # data = hb3_13_and_hb3_11_vs3d7_forPlotting # ) + geom_rect (aes (xmin = col.1 ,xmax = col.2 ,ymin = - 0.40 ,ymax = - 0.25 ,start = start,end = end,length = length, other = other, otherStart = otherStart,otherEnd = otherEnd, color = "Exact Matches \n 3D7 chr11" ,fill = "Exact Matches \n 3D7 chr11" data = hb3_13_and_hb3_11_vs3d7_exacts_forPlotting+ + theme (axis.text.y = element_blank (),axis.ticks.y = element_blank ()) + scale_fill_manual ("Genes \n Description" ,values = geneAnnotationsColors,breaks = names (rawGeneAnnotationsColors),labels = names (rawGeneAnnotationsColors),guide = guide_legend (override.aes = list (color = rawGeneAnnotationsColors,fill = rawGeneAnnotationsColors,alpha = 1 ,shape = 22 ,size = 5 nrow = 8 ,order = 1 + scale_color_manual ("Genomic \n Elements" ,values = geneAnnotationsColors,breaks = names (genomicElementsColors),labels = names (genomicElementsColors),guide = guide_legend (override.aes = list (color = genomicElementsColors,fill = genomicElementsColors,alpha = 1 ,shape = 22 ,size = 5 ncol = 1 ,order = 2 + labs (title = "Annotations of HB3 chr11 and chr13" , fill = "" ) + theme (legend.text = element_text (size = 30 ), axis.text.x = element_text (size = 30 , color = "black" ), plot.title = element_text (size = 30 , color = "black" ), strip.text.y = element_text (size = 30 , color = "black" ), legend.title = element_text (size = 30 , face = "bold" ), legend.box= "vertical" , legend.margin= margin (),legend.background = element_blank (),legend.box.background = element_rect (colour = "black" )) ``` ```{r} #| fig-column: screen #| column: screen ggplotly (hb3_chr11_chr13_annotation_plots)``` ```{r} #| fig-column: screen #| fig-width: 25 #| fig-height: 15 print (hb3_chr11_chr13_annotation_plots)``` ```{r} pdf ("hb3_chr11_chr13_annotation_plots.pdf" , width = 25 , height = 15 , useDingbats = F)print (hb3_chr11_chr13_annotation_plots)dev.off ()```