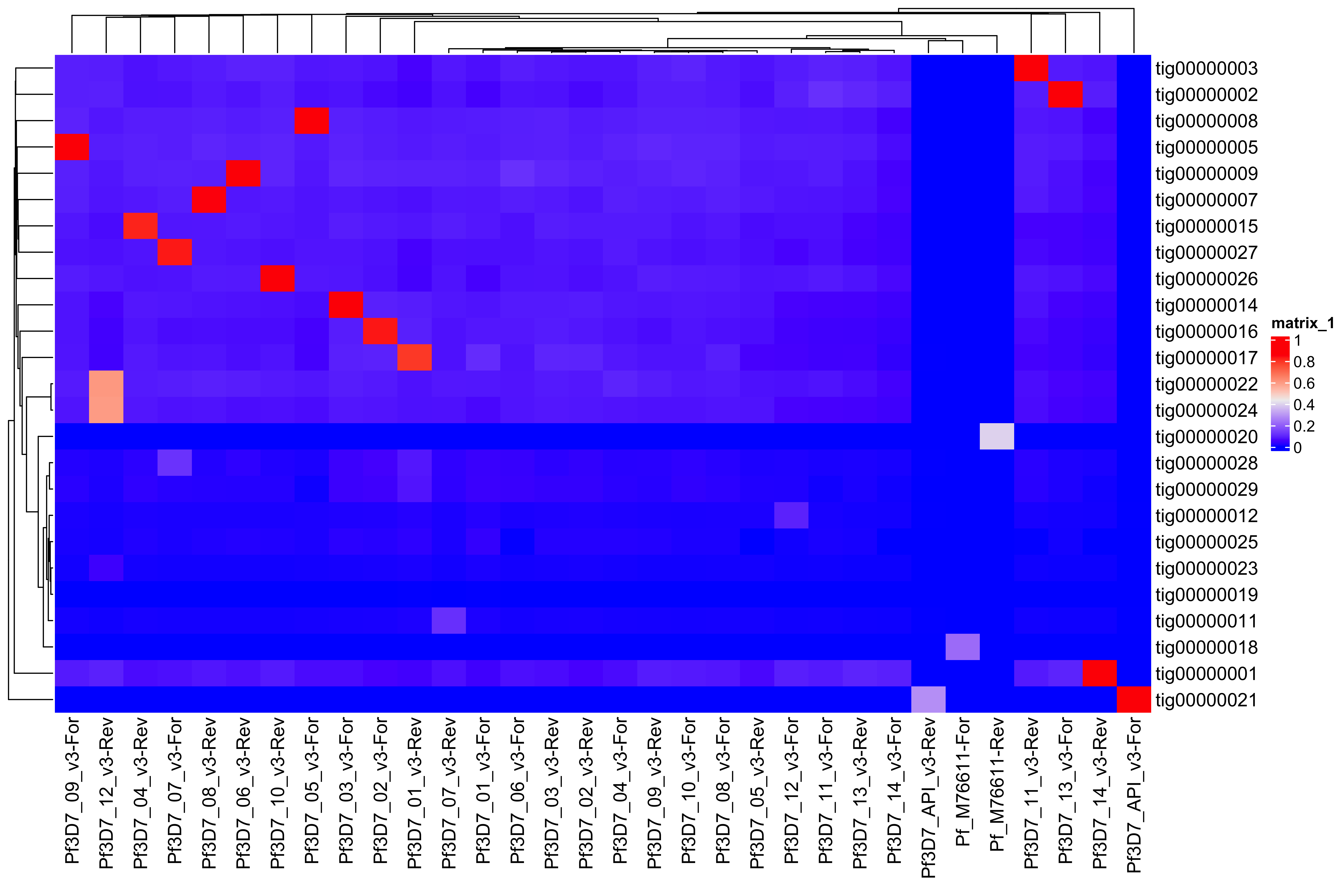
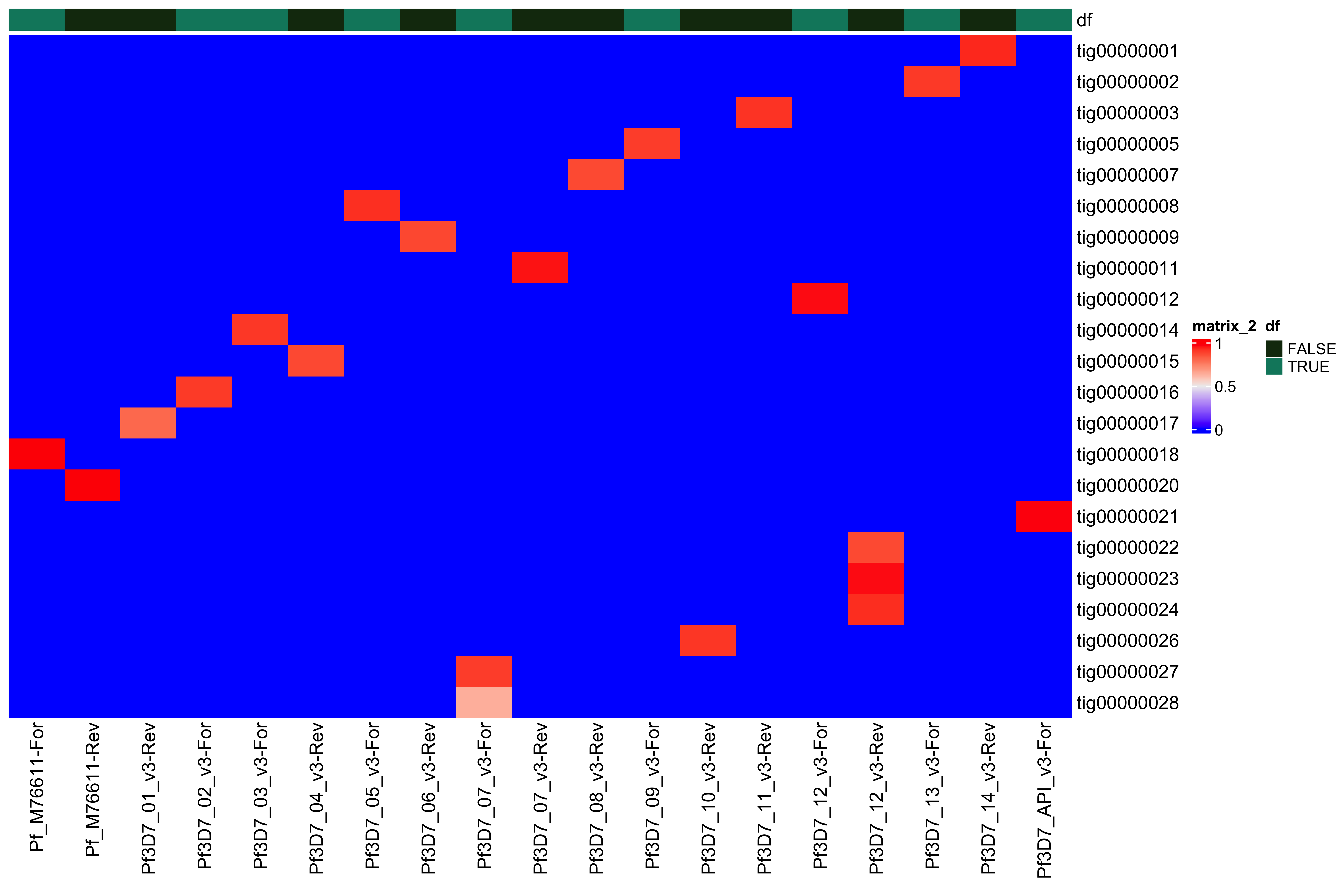
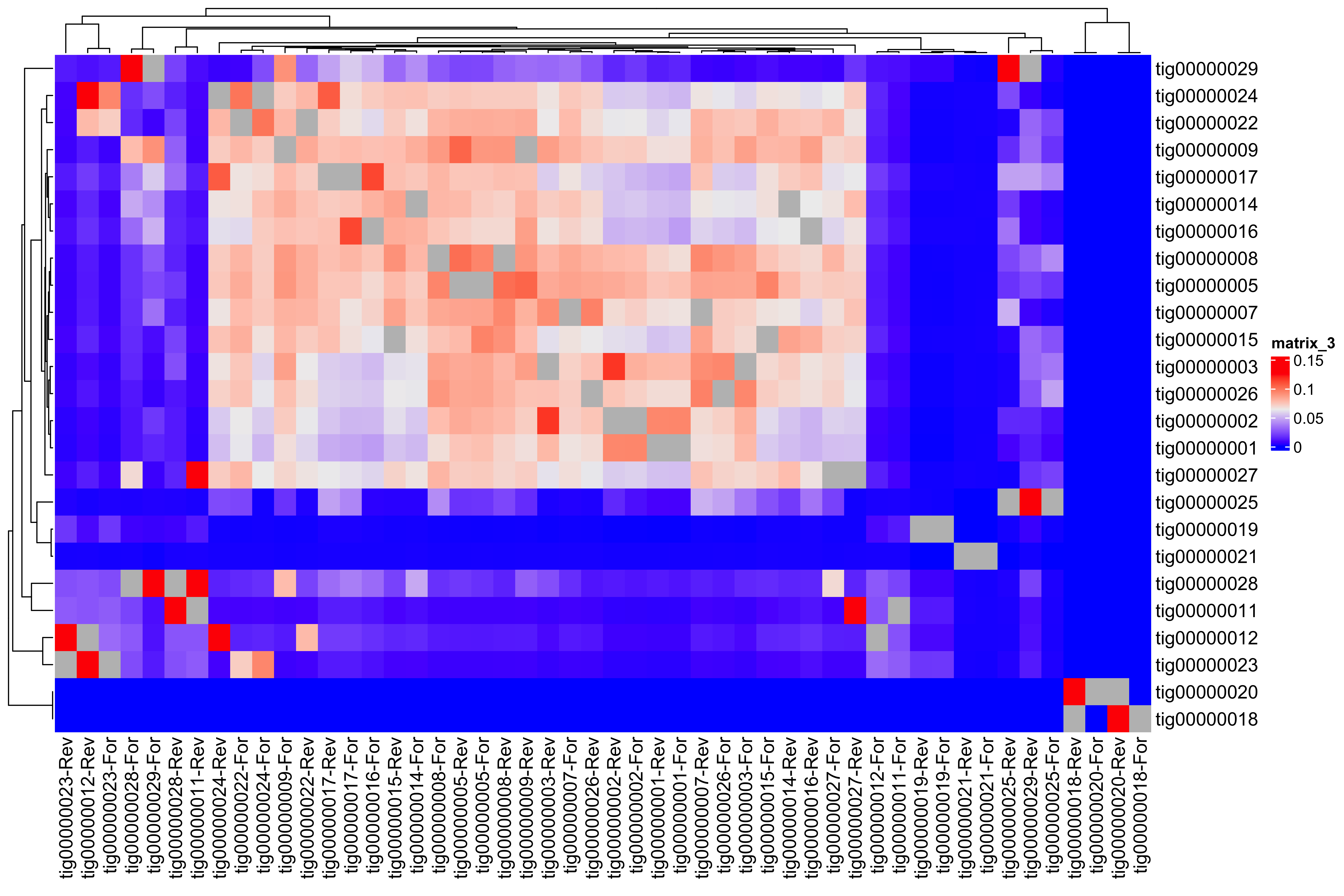
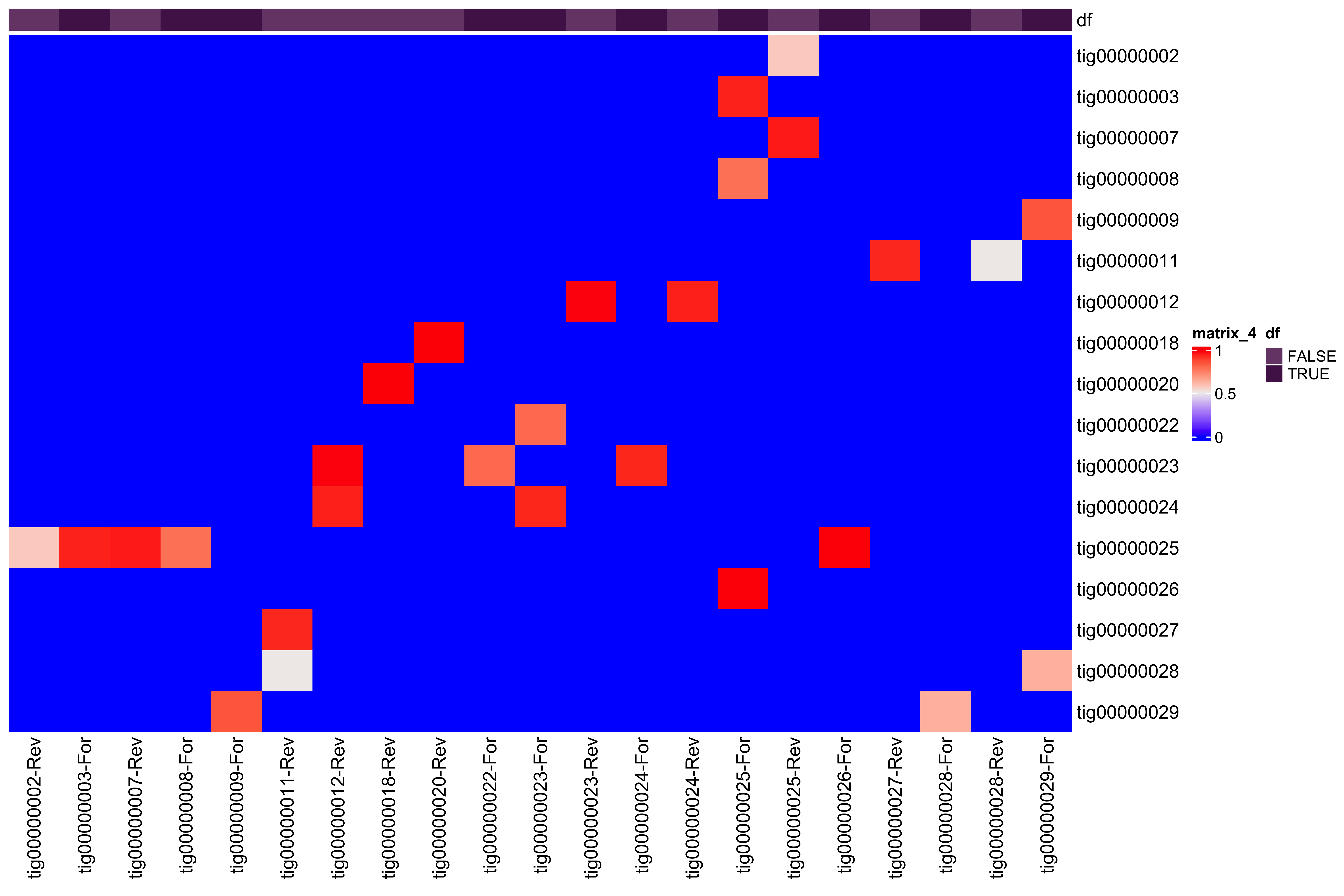
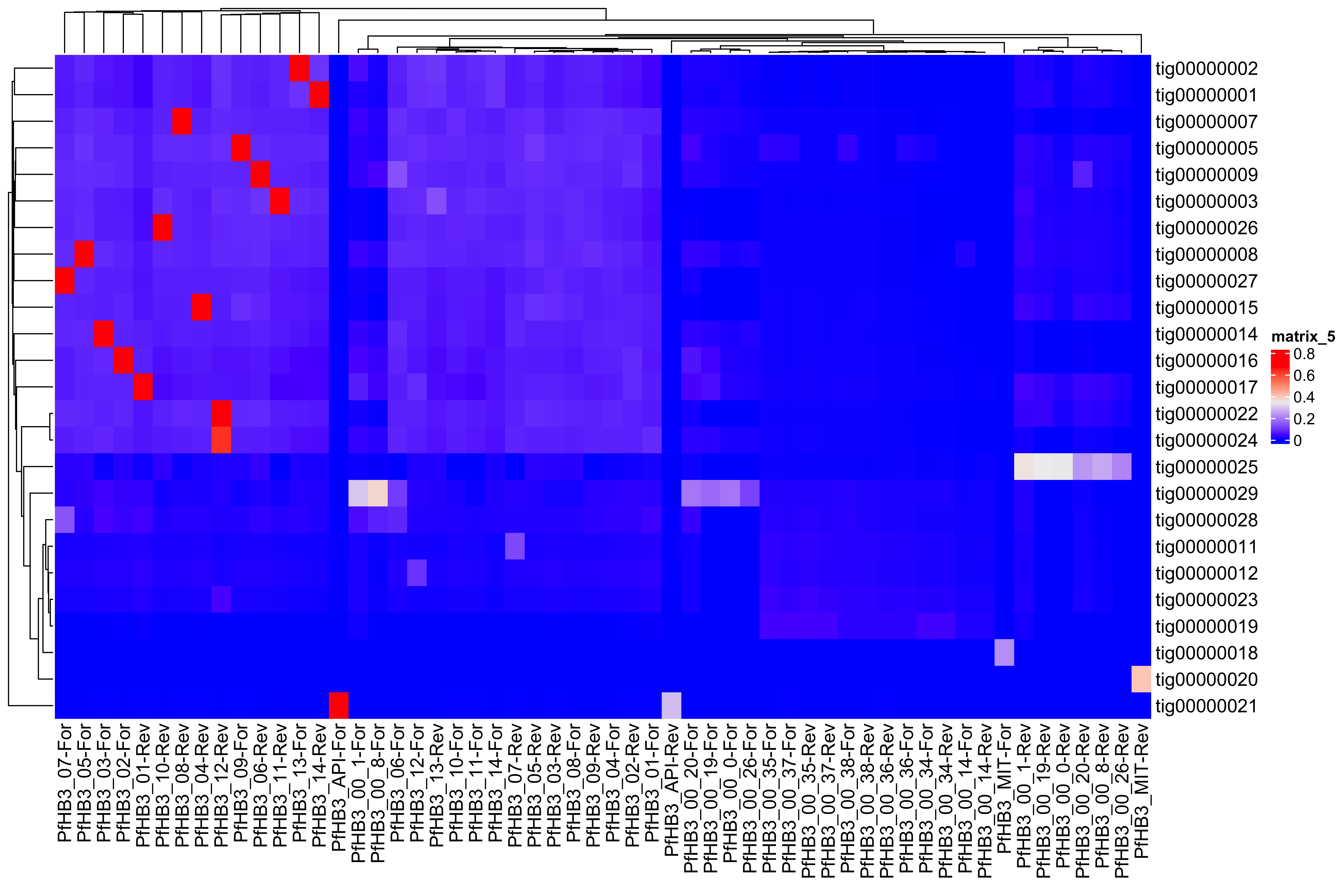
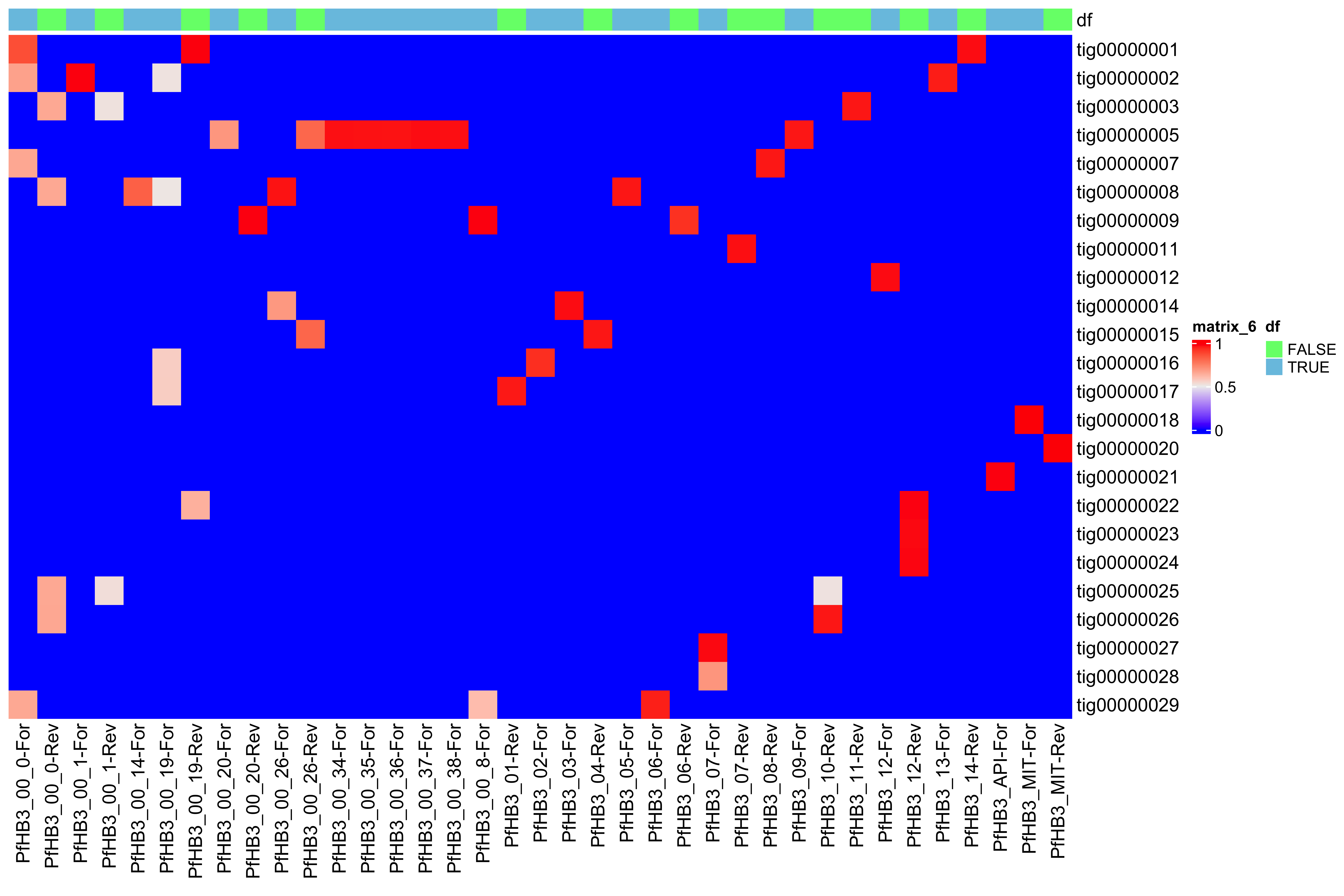
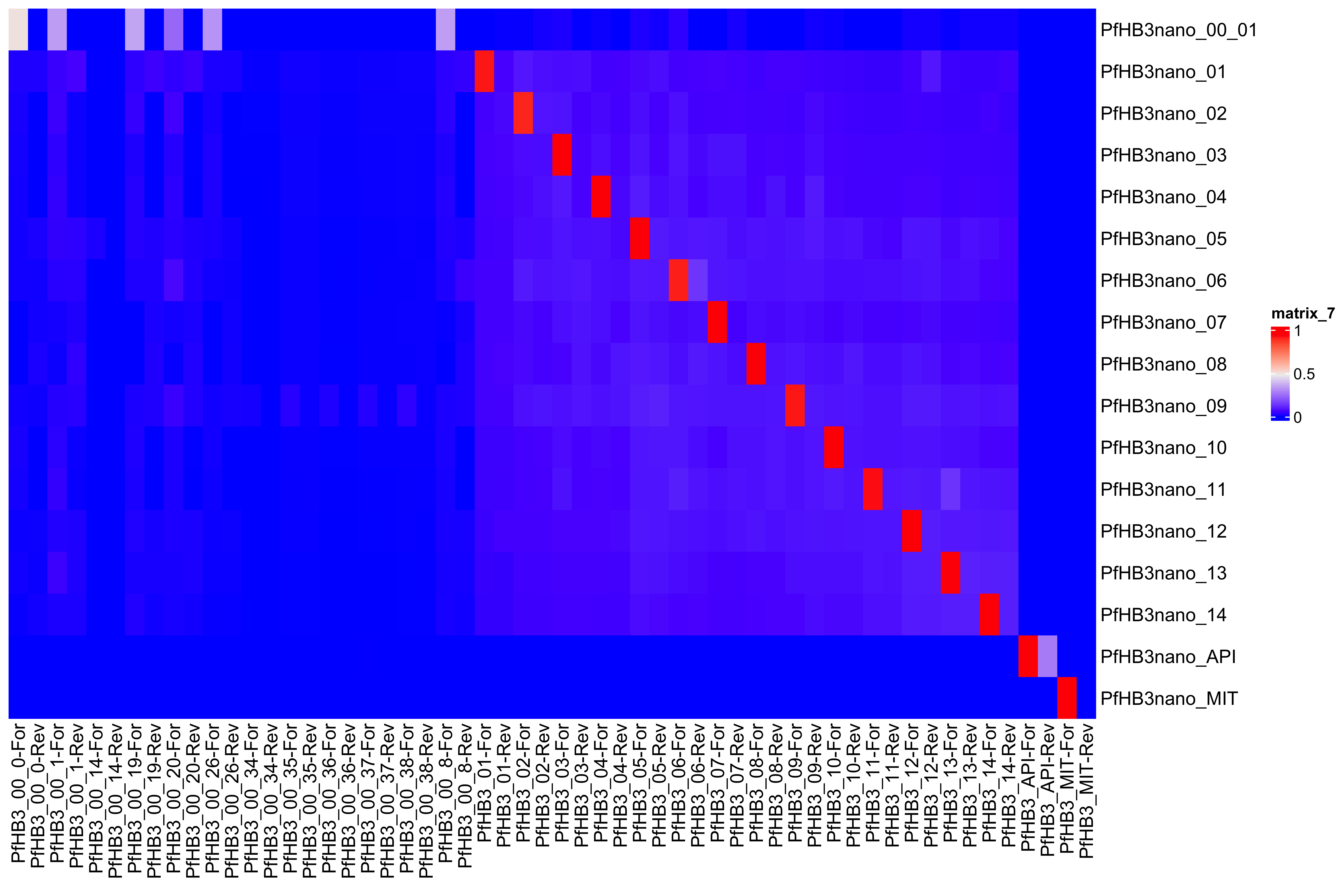
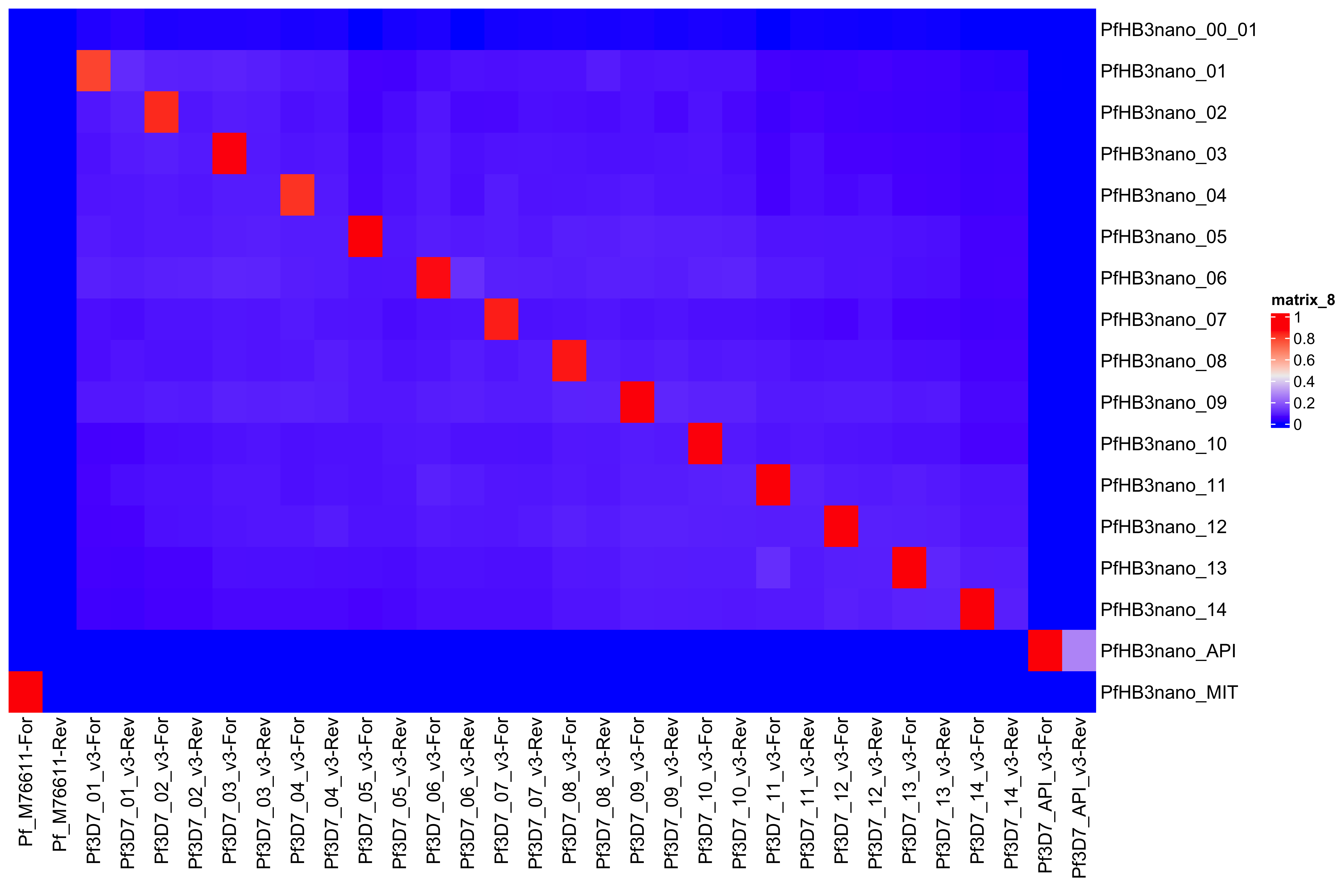
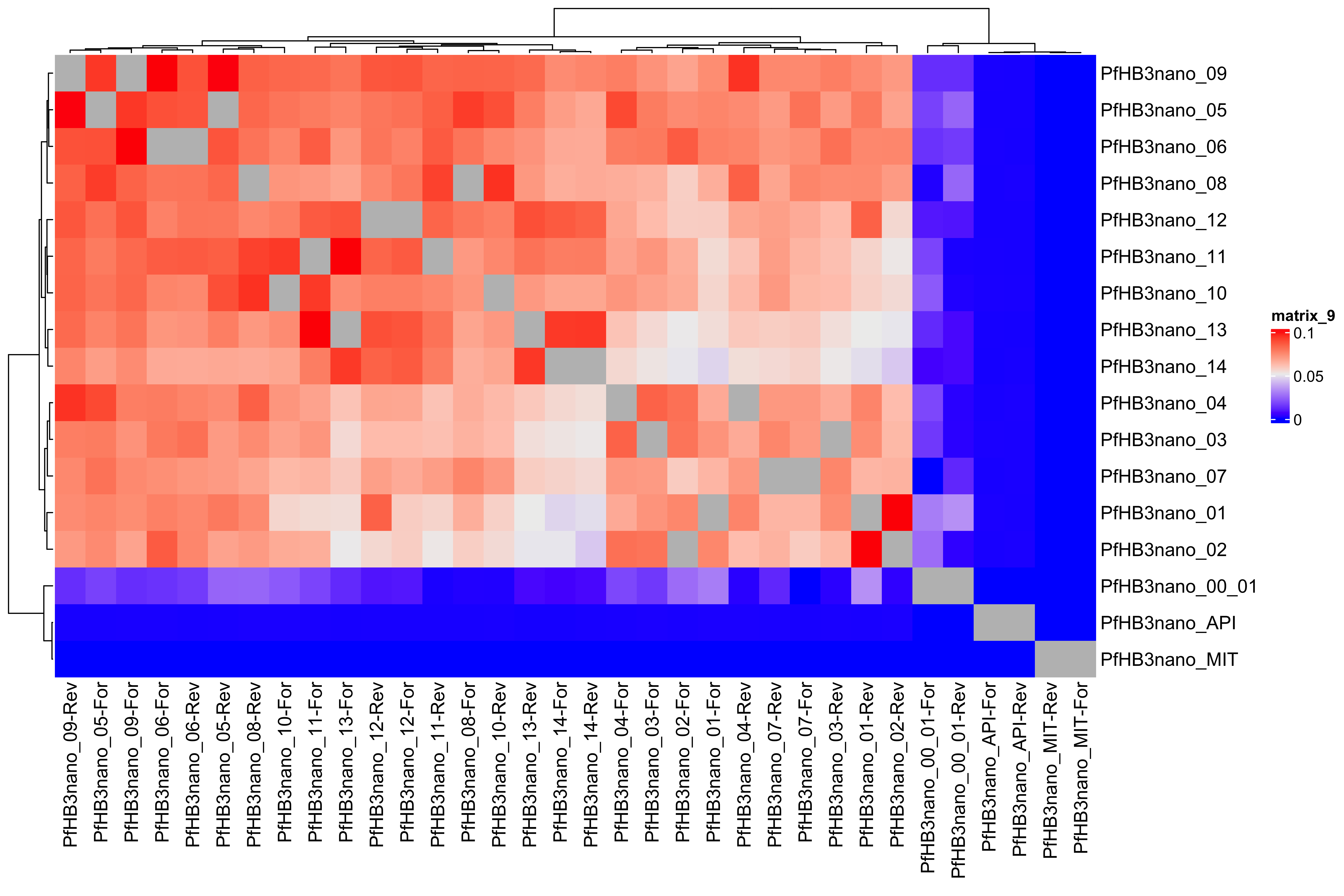
--- title: Full assembly processing of HB3 --- ```{r setup, echo=FALSE, message=FALSE} source("../common.R") ``` ## K-mer comparision ```{bash, eval = F} minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta canuoutput_PfHB3_completegenome.contigs.fasta -a | samtools sort -o canuoutput_PfHB3_completegenome_againstPfHB3.sorted.bam minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta canuoutput_PfHB3_completegenome.contigs.fasta -a | samtools sort -o canuoutput_PfHB3_completegenome_againstPf3D7.sorted.bam nohup elucidator getKmerDetailedKmerDistAgainstRef --fasta canuoutput_PfHB3_completegenome.contigs.fasta --kmerLength 19 --ref /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta --numThreads 48 --overWrite --out against_Pf3D7_klen19.tab.txt --getRevComp & nohup elucidator getKmerDetailedKmerDistAgainstRef --fasta canuoutput_PfHB3_completegenome.contigs.fasta --kmerLength 19 --ref /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta --numThreads 48 --overWrite --out against_PfHB3_klen19.tab.txt --getRevComp & nohup elucidator getKmerDetailedKmerDistAgainstRef --fasta canuoutput_PfHB3_completegenome.contigs.fasta --kmerLength 19 --ref canuoutput_PfHB3_completegenome.contigs.fasta --numThreads 48 --overWrite --out against_self_klen19.tab.txt --getRevComp & ``` ### Against Pf3D7 ```{r} = readr:: read_tsv ("against_Pf3D7_klen19.tab.txt" )= compAgainstPf3D7 %>% mutate (name_short = gsub (" \\ .*" , "" , name)) %>% mutate (ref_short = gsub (" \\ .*" , "" , ref)) %>% mutate (ref_short_orient = paste0 (ref_short, ifelse (revComp, "-Rev" , "-For" )))= compAgainstPf3D7 %>% select (name_short, ref_short_orient, dist)= compAgainstPf3D7_sim %>% spread (ref_short_orient, dist)= as.matrix (compAgainstPf3D7_sim_sp[,2 : ncol (compAgainstPf3D7_sim_sp)])rownames (compAgainstPf3D7_sim_sp_mat) = compAgainstPf3D7_sim_sp$ name_short:: Heatmap (compAgainstPf3D7_sim_sp_mat)= compAgainstPf3D7 %>% arrange (desc (dist))``` ```{r} = compAgainstPf3D7 %>% filter (distLenAdjust > 0.5 ) %>% select (name_short, ref_short_orient, distLenAdjust)= compAgainstPf3D7_sim %>% spread (ref_short_orient, distLenAdjust, fill = 0 )= as.matrix (compAgainstPf3D7_sim_sp[,2 : ncol (compAgainstPf3D7_sim_sp)])rownames (compAgainstPf3D7_sim_sp_mat) = compAgainstPf3D7_sim_sp$ name_short= tibble (name = colnames (compAgainstPf3D7_sim_sp_mat)) %>% mutate (plusStrand = grepl ("-For" ,name)) %>% as.data.frame ()= ComplexHeatmap:: HeatmapAnnotation (df = topDf[,c ("plusStrand" )]):: Heatmap (compAgainstPf3D7_sim_sp_mat, cluster_rows = F, cluster_columns = F, top_annotation = topAnno )``` ```{r} = compAgainstPf3D7 %>% filter (distLenAdjust > 0.5 ) ``` ```{r} = compAgainstPf3D7 %>% group_by (name) %>% filter (distLenAdjust == max (distLenAdjust))``` ### Against Self ```{r} = readr:: read_tsv ("against_self_klen19.tab.txt" ) %>% filter (name != ref)= compAgainstself %>% mutate (name_short = gsub (" \\ .*" , "" , name)) %>% mutate (ref_short = gsub (" \\ .*" , "" , ref)) %>% mutate (ref_short_orient = paste0 (ref_short, ifelse (revComp, "-Rev" , "-For" )))= compAgainstself %>% select (name_short, ref_short_orient, dist)= compAgainstself_sim %>% spread (ref_short_orient, dist)= as.matrix (compAgainstself_sim_sp[,2 : ncol (compAgainstself_sim_sp)])rownames (compAgainstself_sim_sp_mat) = compAgainstself_sim_sp$ name_short:: Heatmap (compAgainstself_sim_sp_mat)= compAgainstself %>% arrange (desc (dist))``` ```{r} = compAgainstself %>% filter (distLenAdjust > 0.5 ) %>% select (name_short, ref_short_orient, distLenAdjust)= compAgainstself_sim %>% spread (ref_short_orient, distLenAdjust, fill = 0 )= as.matrix (compAgainstself_sim_sp[,2 : ncol (compAgainstself_sim_sp)])rownames (compAgainstself_sim_sp_mat) = compAgainstself_sim_sp$ name_short= tibble (name = colnames (compAgainstself_sim_sp_mat)) %>% mutate (plusStrand = grepl ("-For" ,name)) %>% as.data.frame ()= ComplexHeatmap:: HeatmapAnnotation (df = topDf[,c ("plusStrand" )]):: Heatmap (compAgainstself_sim_sp_mat, cluster_rows = F, cluster_columns = F, top_annotation = topAnno )``` ```{r} = compAgainstself %>% filter (distLenAdjust > 0.5 ) = compAgainstself %>% group_by (name) %>% filter (totalKmersIn2 > totalKmersIn1) %>% filter (distLenAdjust == max (distLenAdjust)) %>% filter (distLenAdjust > 0.9 )``` ### Against previous PfHB3 ```{r} = readr:: read_tsv ("against_PfHB3_klen19.tab.txt" )= compAgainstPfHB3 %>% mutate (name_short = gsub (" \\ .*" , "" , name)) %>% mutate (ref_short = gsub (" \\ .*" , "" , ref)) %>% mutate (ref_short_orient = paste0 (ref_short, ifelse (revComp, "-Rev" , "-For" )))= compAgainstPfHB3 %>% select (name_short, ref_short_orient, dist)= compAgainstPfHB3_sim %>% spread (ref_short_orient, dist)= as.matrix (compAgainstPfHB3_sim_sp[,2 : ncol (compAgainstPfHB3_sim_sp)])rownames (compAgainstPfHB3_sim_sp_mat) = compAgainstPfHB3_sim_sp$ name_short:: Heatmap (compAgainstPfHB3_sim_sp_mat)= compAgainstPfHB3 %>% arrange (desc (dist))``` ```{r} = compAgainstPfHB3 %>% filter (distLenAdjust > 0.5 ) %>% select (name_short, ref_short_orient, distLenAdjust)= compAgainstPfHB3_sim %>% spread (ref_short_orient, distLenAdjust, fill = 0 )= as.matrix (compAgainstPfHB3_sim_sp[,2 : ncol (compAgainstPfHB3_sim_sp)])rownames (compAgainstPfHB3_sim_sp_mat) = compAgainstPfHB3_sim_sp$ name_short= tibble (name = colnames (compAgainstPfHB3_sim_sp_mat)) %>% mutate (plusStrand = grepl ("-For" ,name)) %>% as.data.frame ()= ComplexHeatmap:: HeatmapAnnotation (df = topDf[,c ("plusStrand" )]):: Heatmap (compAgainstPfHB3_sim_sp_mat, cluster_rows = F, cluster_columns = F, top_annotation = topAnno )``` ```{r} = compAgainstPfHB3 %>% filter (distLenAdjust > 0.5 ) = compAgainstPfHB3_sim_filt %>% group_by (name) %>% filter (dist == max (dist))= compAgainstPfHB3 %>% group_by (name) %>% filter (! grepl ("_00" , ref)) %>% filter (distLenAdjust == max (distLenAdjust))``` ## Finding best fits ```{r} = compAgainstPfHB3_bestFit %>% group_by () %>% #select(name, ref, name_short, ref_short, revComp) %>% mutate (genome = "PfHB3" ) %>% bind_rows (compAgainstPf3D7_bestFit %>% group_by () %>% #select(name, ref, name_short, ref_short, revComp) %>% mutate (genome = "Pf3D7" )) %>% filter (name %!in% compAgainstself_bestLenAdjust$ name) %>% filter (totalKmersIn1 > 30000 ) # recommend finding where tig00000029 overlaps with tig00000009 (chr6) and then take the left # recommend finding where tig00000028 overlaps with tig00000027 (chr7) and then take the left # recommend finding where tig00000022 overlaps with tig00000024 (chr12) = bestFits %>% mutate (chrom = gsub ("PfHB3_" , "" , ref_short))%>% mutate (chrom = gsub ("Pf3D7_" , "" , chrom))%>% mutate (chrom = gsub ("_v3" , "" , chrom)) %>% mutate (chrom = gsub ("Pf_M76611" , "MIT" , chrom)) # manual renames = c ("tig00000029" , "tig00000028" , "tig00000022" )= bestFits %>% filter (name_short %!in% toRemoved) %>% select (name, chrom, revComp) %>% unique () %>% arrange (chrom) %>% group_by (chrom) %>% arrange (desc (name)) %>% mutate (row = row_number (), n = n ()) %>% mutate (newName = paste0 ("PfHB3nano_" , chrom)) %>% mutate (newName = ifelse (n > 1 , paste0 (newName, "_" , row), newName)) %>% arrange (chrom)%>% mutate (name_short = gsub (" \\ .*" , "" , name)) %>% mutate (name = ifelse (revComp, paste0 (name, "_Comp" ), name ) )write_tsv (bestFits_output %>% group_by () %>% select (name, newName) %>% bind_rows (tibble (name = "tig00000029 len=77951 reads=109 class=contig suggestRepeat=no suggestBubble=no suggestCircular=no trim=0-77951" ,newName = "PfHB3nano_00_01" )# ) %>% # bind_rows( # tibble(name = "tig00000028 len=147642 reads=355 class=contig suggestRepeat=no suggestBubble=no suggestCircular=no trim=0-147642", # newName = "PfHB3nano_00_02") col_names = F, "renaming_file.txt" )write_tsv (bestFits_output %>% group_by ()%>% filter (chrom %!in% c ("07" , "12" , "MIT" , "API" )) %>% select (name) , col_names = F, "extracting_file.txt" )``` ```{bash, eval = F} # reorient contigs to be the direction of the 3D7 chromosomes to keep consistency elucidator reOrientReads --reOrientToBestWinner --fasta canuoutput_PfHB3_completegenome.contigs.fasta --ref /tank/data/genomes/plasmodium//genomes/pf/genomes/PfHB3.fasta --kLength 11 --overWrite --numThreads 40 elucidator faToTwoBit --in reOriented_canuoutput_PfHB3_completegenome.contigs.fasta --overWrite --out reOriented_canuoutput_PfHB3_completegenome.contigs.2bit elucidator chromVsChromUniqueComp --genome2bit reOriented_canuoutput_PfHB3_completegenome.contigs.2bit --out nanoHB3_comp_klen31_revComp --checkComplement --overWrite --numThreads 48 elucidator chromVsChromUniqueComp --genome2bit reOriented_canuoutput_PfHB3_completegenome.contigs.2bit --out tight_nanoHB3_comp_klen31_revComp --minDisForGrouping 300 --checkComplement --overWrite --numThreads 48 elucidator bedCoordSort --bed tight_nanoHB3_comp_klen31_revComp_groupRegions.bed | elucidator filterBedRecordsByLength --minLen 10000 --bed STDIN > filt_tight_nanoHB3_comp_klen31_revComp_groupRegions.bed ``` ```{r} = readr:: read_tsv ("filt_tight_nanoHB3_comp_klen31_revComp_groupRegions.bed" , col_names = F) %>% separate (X4, into = c ("chrom1_section" , "chrom2_section" ), remove = F, sep = "--" ) %>% separate (chrom1_section,into = c ("chrom1_section_chrom" , "chrom1_section_start" , "chrom1_section_end" , "chrom1_section_strand" ),remove = F, sep = "-" , convert = T) %>% separate (chrom2_section,into = c ("chrom2_section_chrom" , "chrom2_section_start" , "chrom2_section_end" , "chrom2_section_strand" ),remove = F, sep = "-" , convert = T) %>% mutate (chrom2_section_len = chrom2_section_end - chrom2_section_start, chrom1_section_len = chrom1_section_end - chrom1_section_start)= filt_tight_nanoHB3_comp_klen31_revComp_groupRegions %>% filter (X1 %in% bestFits_output$ name_short,chrom1_section_chrom %in% bestFits_output$ name_short, chrom2_section_chrom %in% bestFits_output$ name_short) = filt_tight_nanoHB3_comp_klen31_revComp_groupRegions_filt %>% filter ("tig00000022" == X1 | "tig00000024" == X1)``` # Trimming and renaming ## Chromosomal ```{bash, eval = F} # chr06 # trim tig00000029 egrep tig00000029 -A 1 reOriented_canuoutput_PfHB3_completegenome.contigs.fasta > tig00000029.fasta elucidator trimFront --forwardBases 56237 --fasta tig00000029.fasta --out trimFront_56237_tig00000029.fasta --overWrite ## combine with tig00000009 egrep tig00000009 -A 1 reOriented_canuoutput_PfHB3_completegenome.contigs.fasta > tig00000009.fasta elucidator appendReads --fasta tig00000009.fasta --seq trimFront_56237_tig00000029.fasta --overWrite minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta appended_tig00000009.fasta -a | samtools sort -o appended_tig00000009_againstPfHB3.sorted.bam minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta appended_tig00000009.fasta -a | samtools sort -o appended_tig00000009_againstPf3D7.sorted.bam minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta tig00000009.fasta -a | samtools sort -o tig00000009_againstPfHB3.sorted.bam minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta tig00000009.fasta -a | samtools sort -o tig00000009_againstPf3D7.sorted.bam minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta trimFront_56237_tig00000029.fasta -a | samtools sort -o trimFront_56237_tig00000029_againstPfHB3.sorted.bam minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta trimFront_56237_tig00000029.fasta -a | samtools sort -o trimFront_56237_tig00000029_againstPf3D7.sorted.bam minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta tig00000029.fasta -a | samtools sort -o tig00000029_againstPf3D7.sorted.bam minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta tig00000029.fasta -a | samtools sort -o tig00000029_againstPfHB3.sorted.bam minimap2 tig00000009.fasta tig00000029.fasta -a | samtools sort -o tig00000029_againsttig00000009.sorted.bam minimap2 tig00000009.fasta trimFront_56237_tig00000029.fasta -a | samtools sort -o trimFront_56237_tig00000029_againsttig00000009.sorted.bam # though these appear very similar enough to have thought they should stitch together, they both end with the telomeric end repeat of Pf so likely tig00000009 goes to the end of the chromosome # chr07 # trim tig00000028 egrep tig00000028 -A 1 reOriented_canuoutput_PfHB3_completegenome.contigs.fasta > tig00000028.fasta elucidator trimFront --forwardBases 41931 --fasta tig00000028.fasta --out trimFront_41931_tig00000028.fasta --overWrite elucidator trimToLen --length 51066 --fasta trimFront_41931_tig00000028.fasta --overWrite --out trimToLen_51066_trimFront_41931_tig00000028.fasta ## combine with tig00000027 egrep tig00000027 -A 1 reOriented_canuoutput_PfHB3_completegenome.contigs.fasta > tig00000027.fasta elucidator appendReads --fasta tig00000027.fasta --seq trimToLen_51066_trimFront_41931_tig00000028.fasta --overWrite minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta appended_tig00000027.fasta -a | samtools sort -o appended_tig00000027_againstPfHB3.sorted.bam minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta tig00000027.fasta -a | samtools sort -o tig00000027_againstPfHB3.sorted.bam # was originally left separate, but post analysis reveals most likely this portion is truly chr 7 end # chr 12 # combine tig00000022 and tig00000024 egrep tig00000022 -A 1 reOriented_canuoutput_PfHB3_completegenome.contigs.fasta > tig00000022.fasta elucidator trimFront --forwardBases 42888 --fasta tig00000022.fasta --out trimFront_42888_tig00000022.fasta --overWrite egrep tig00000024 -A 1 reOriented_canuoutput_PfHB3_completegenome.contigs.fasta > tig00000024.fasta elucidator appendReads --fasta tig00000024.fasta --seq trimFront_42888_tig00000022.fasta --overWrite minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta appended_tig00000024.fasta -a | samtools sort -o appended_tig00000024_againstPfHB3.sorted.bam ``` ## Mitochondrion and Apicoplast ### Mitochondrion ```{bash, eval = F} # trim MIT tig00000018 egrep Pf_M76611 -A1 /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta > Pf_M76611.fasta egrep tig00000018 -A 1 reOriented_canuoutput_PfHB3_completegenome.contigs.fasta > tig00000018.fasta elucidator reOrientCirculateGenomeToRef --fasta tig00000018.fasta --ref Pf_M76611.fasta minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta trimmed_tig00000018.fasta -a | samtools sort -o trimmed_tig00000018_againstPfHB3.sorted.bam ``` ### Apicoplast ```{bash, eval = F} # trim API tig00000021 egrep Pf3D7_API_v3 -A1 /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta > Pf3D7_API_v3.fasta egrep tig00000021 -A 1 reOriented_canuoutput_PfHB3_completegenome.contigs.fasta > tig00000021.fasta elucidator reOrientCirculateGenomeToRef --fasta tig00000021.fasta --ref Pf3D7_API_v3.fasta minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta trimmed_tig00000021.fasta -a | samtools sort -o trimmed_tig00000021_againstPfHB3.sorted.bam ``` ## Combining ```{bash, eval = F} elucidator extractByName --names extracting_file.txt --fasta reOriented_canuoutput_PfHB3_completegenome.contigs.fasta --overWrite cat trimFront_56237_tig00000029.fasta trimmed_tig00000018.fasta trimmed_tig00000021.fasta appended_tig00000024.fasta appended_tig00000027.fasta extracted_reOriented_canuoutput_PfHB3_completegenome.contigs.fasta > raw_combined.fasta elucidator renameIDs --keyIn renaming_file.txt --fasta raw_combined.fasta --overWrite elucidator sortReads --sortBy name --fasta renamed_raw_combined.fasta --overWrite --out PfHB3nano.fasta elucidator getReadLens --addHeader chrom,length --fasta PfHB3nano.fasta --overWrite --out PfHB3nano_chrom_length.tab.txt elucidator faToTwoBit --in PfHB3nano.fasta --out PfHB3nano.2bit --overWrite ``` # Final checks ```{bash, eval = F} # checks #kmer dist to Pf3D7, PfHB3, and minimaps minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta PfHB3nano.fasta -a | samtools sort -o PfHB3nano_againstPfHB3.sorted.bam minimap2 /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta PfHB3nano.fasta -a | samtools sort -o PfHB3nano_againstPf3D7.sorted.bam nohup elucidator getKmerDetailedKmerDistAgainstRef --fasta PfHB3nano.fasta --kmerLength 19 --ref /tank/data/genomes/plasmodium/genomes/pf/genomes/Pf3D7.fasta --numThreads 48 --overWrite --out PfHB3nano_against_Pf3D7_klen19.tab.txt --getRevComp & nohup elucidator getKmerDetailedKmerDistAgainstRef --fasta PfHB3nano.fasta --kmerLength 19 --ref /tank/data/genomes/plasmodium/genomes/pf/genomes/PfHB3.fasta --numThreads 48 --overWrite --out PfHB3nano_against_PfHB3_klen19.tab.txt --getRevComp & nohup elucidator getKmerDetailedKmerDistAgainstRef --fasta PfHB3nano.fasta --kmerLength 19 --ref PfHB3nano.fasta --numThreads 48 --overWrite --out PfHB3nano_against_self_klen19.tab.txt --getRevComp & nohup elucidator chromVsChromUniqueComp --genome2bit PfHB3nano.2bit --out PfHB3nano_comp_klen31_revComp --minDisForGrouping 300 --checkComplement --overWrite --numThreads 48 & ``` ## Heatmaps ### Against HB3 ```{r} = readr:: read_tsv ("PfHB3nano_against_PfHB3_klen19.tab.txt" )= compAgainstPfHB3 %>% mutate (name_short = gsub (" \\ .*" , "" , name)) %>% mutate (ref_short = gsub (" \\ .*" , "" , ref)) %>% mutate (ref_short_orient = paste0 (ref_short, ifelse (revComp, "-Rev" , "-For" )))= compAgainstPfHB3 %>% select (name_short, ref_short_orient, dist)= compAgainstPfHB3_sim %>% spread (ref_short_orient, dist)= as.matrix (compAgainstPfHB3_sim_sp[,2 : ncol (compAgainstPfHB3_sim_sp)])rownames (compAgainstPfHB3_sim_sp_mat) = compAgainstPfHB3_sim_sp$ name_short:: Heatmap (compAgainstPfHB3_sim_sp_mat, cluster_rows = F, cluster_columns = F)``` ### Against 3D7 ```{r} = readr:: read_tsv ("PfHB3nano_against_Pf3D7_klen19.tab.txt" )= compAgainstPf3D7 %>% mutate (name_short = gsub (" \\ .*" , "" , name)) %>% mutate (ref_short = gsub (" \\ .*" , "" , ref)) %>% mutate (ref_short_orient = paste0 (ref_short, ifelse (revComp, "-Rev" , "-For" )))= compAgainstPf3D7 %>% select (name_short, ref_short_orient, dist)= compAgainstPf3D7_sim %>% spread (ref_short_orient, dist)= as.matrix (compAgainstPf3D7_sim_sp[,2 : ncol (compAgainstPf3D7_sim_sp)])rownames (compAgainstPf3D7_sim_sp_mat) = compAgainstPf3D7_sim_sp$ name_short:: Heatmap (compAgainstPf3D7_sim_sp_mat, cluster_rows = F, cluster_columns = F)``` ### Against Self ```{r} = readr:: read_tsv ("PfHB3nano_against_self_klen19.tab.txt" )= compAgainstSelf %>% filter (name != ref) %>% mutate (name_short = gsub (" \\ .*" , "" , name)) %>% mutate (ref_short = gsub (" \\ .*" , "" , ref)) %>% mutate (ref_short_orient = paste0 (ref_short, ifelse (revComp, "-Rev" , "-For" )))= compAgainstSelf %>% select (name_short, ref_short_orient, dist)= compAgainstSelf_sim %>% spread (ref_short_orient, dist)= as.matrix (compAgainstSelf_sim_sp[,2 : ncol (compAgainstSelf_sim_sp)])rownames (compAgainstSelf_sim_sp_mat) = compAgainstSelf_sim_sp$ name_short:: Heatmap (compAgainstSelf_sim_sp_mat) ``` ### Long homology ```{r} = readr:: read_tsv ("PfHB3nano_chrom_length.tab.txt" )``` ```{r} #pf3d7Genes = readr::read_tsv("Pf3D7_genes.tab.txt") ``` ```{r} = readr:: read_tsv ("PfHB3nano_comp_klen31_revComp.tab.txt" ) %>% mutate (pair = paste0 (chrom1, "--vs--" , chrom2))= conservedInfo %>% separate (group, into = c ("chrom1_section" , "chrom2_section" ), remove = F, sep = "--" ) %>% separate (chrom1_section,into = c ("chrom1_section_chrom" , "chrom1_section_start" , "chrom1_section_end" , "chrom1_section_strand" ),remove = F, sep = "-" , convert = T) %>% separate (chrom2_section,into = c ("chrom2_section_chrom" , "chrom2_section_start" , "chrom2_section_end" , "chrom2_section_strand" ),remove = F, sep = "-" , convert = T) %>% mutate (chrom2_section_len = chrom2_section_end - chrom2_section_start, chrom1_section_len = chrom1_section_end - chrom1_section_start) %>% group_by (group) %>% mutate (totalBases = sum (size)) %>% arrange (desc (totalBases)) %>% mutate (chrom1Pos_start = ifelse (chrom1RevComp, chrom1Pos + size, chrom1Pos), crhom1Pos_end = ifelse (chrom1RevComp, chrom1Pos, chrom1Pos + size)) %>% mutate (chrom1_fracConserved = totalBases/ chrom1_section_len, chrom2_fracConserved = totalBases/ chrom2_section_len)= conservedInfo_mod %>% select (group, totalBases, starts_with ("chrom1_" ), starts_with ("chrom2_" )) %>% unique ():: datatable (conservedInfo_mod_sel,extensions = 'Buttons' , options = list (dom = 'Bfrtip' ,buttons = c ('csv' )= list ()for (grouping in unique (conservedInfo_mod$ group)[1 : 10 ]){= conservedInfo_mod %>% filter (grouping == group)# pf3d7Genes_chrom1 = pf3d7Genes %>% # filter(col.0 == conservedInfo_current$chrom1[1], # col.1 >= conservedInfo_current$chrom1_section_start[1] | col.2 >= conservedInfo_current$chrom1_section_start[1], # col.1 <= conservedInfo_current$chrom1_section_end[1] | col.2 <= conservedInfo_current$chrom1_section_end[1]) # # pf3d7Genes_chrom2 = pf3d7Genes %>% # filter(col.0 == conservedInfo_current$chrom2[1], # col.1 >= conservedInfo_current$chrom2_section_start[1] | col.2 >= conservedInfo_current$chrom2_section_start[1], # col.1 <= conservedInfo_current$chrom2_section_end[1] | col.2 <= conservedInfo_current$chrom2_section_end[1]) # #cat(c(grouping, unique(conservedInfo_current$chrom1_section_len)), sep = "\n") = mean (c (conservedInfo_current$ chrom1_section_len, conservedInfo_current$ chrom2_section_len))= ggplotly (ggplot (conservedInfo_current) + geom_segment (aes (x = chrom1Pos_start,xend = crhom1Pos_end - 1 ,y = chrom2Pos,yend = chrom2Pos + size - 1 ,color = chrom1RevComp+ # geom_rect(data = pf3d7Genes_chrom1, # aes(xmin = col.1, xmax = col.2, # ymax = conservedInfo_current$chrom2_section_start[1] - conservedInfo_current$chrom2_section_len[1] * 0.001, # ymin = conservedInfo_current$chrom2_section_start[1] - conservedInfo_current$chrom2_section_len[1] * 0.01, # fill = description, # id = id)) + # geom_rect(data = pf3d7Genes_chrom2, # aes(ymin = col.1, ymax = col.2, # xmax = conservedInfo_current$chrom1_section_start[1] - conservedInfo_current$chrom1_section_len[1] * 0.001, # xmin = conservedInfo_current$chrom1_section_start[1] - conservedInfo_current$chrom1_section_len[1] * 0.01, # fill = description, # id = id )) + labs (title = paste0 (grouping, " \n " , "size=" , size)) + sofonias_theme + coord_equal () + scale_color_manual (values = c ("TRUE" = "#AA0A3C" , "FALSE" = "#0AB45A" ))= PfHB3nanoChroms %>% filter (chrom %in% unique (c (conservedInfo_current$ chrom1, conservedInfo_current$ chrom2)))= PfHB3nanoChroms_filt %>% mutate (chrom = factor (chrom, levels = c (.$ chrom)))= conservedInfo_current %>% group_by (chrom1_section_chrom, chrom1_section_start, chrom1_section_end, %>% summarise (total = n (), revCompSum = sum (chrom1RevComp)) %>% mutate (chrom1RevCompFrac = revCompSum/ total) %>% mutate (chrom1RevComp = ifelse (chrom1RevCompFrac > 0.5 , T, F))= conservedInfo_current_sum %>% mutate (chrom1_section_chrom = factor (chrom1_section_chrom, levels = c (PfHB3nanoChroms_filt$ chrom))) %>% mutate (chrom2_section_chrom = factor (chrom2_section_chrom, levels = c (PfHB3nanoChroms_filt$ chrom)))= unique (conservedInfo_current$ totalBases)paste0 (grouping, "-fullView" )]] = ggplotly (ggplot () + geom_rect (data = PfHB3nanoChroms_filt,aes (xmin = 0 ,xmax = length,ymin = as.numeric (chrom) - 0.4 ,ymax = as.numeric (chrom) + 0.4 fill = "grey75" + scale_y_continuous (breaks = 1 : (nrow (PfHB3nanoChroms_filt)),labels = levels (PfHB3nanoChroms_filt$ chrom)+ + geom_rect (data = conservedInfo_current_sum,aes (xmin = chrom1_section_start,xmax = chrom1_section_end,ymin = as.numeric (chrom1_section_chrom) - 0.4 ,ymax = as.numeric (chrom1_section_chrom) + 0.4 ,fill = chrom1RevComp+ geom_rect (data = conservedInfo_current_sum,aes (xmin = chrom2_section_start,xmax = chrom2_section_end,ymin = as.numeric (chrom2_section_chrom) - 0.4 ,ymax = as.numeric (chrom2_section_chrom) + 0.4 fill = "#0AB45A" + scale_fill_manual (values = c ("TRUE" = "#AA0A3C" , "FALSE" = "#0AB45A" )):: tagList (listOfPlots)= conservedInfo %>% group_by (pair) %>% summarise (total = sum (size)) %>% arrange (desc (total))```