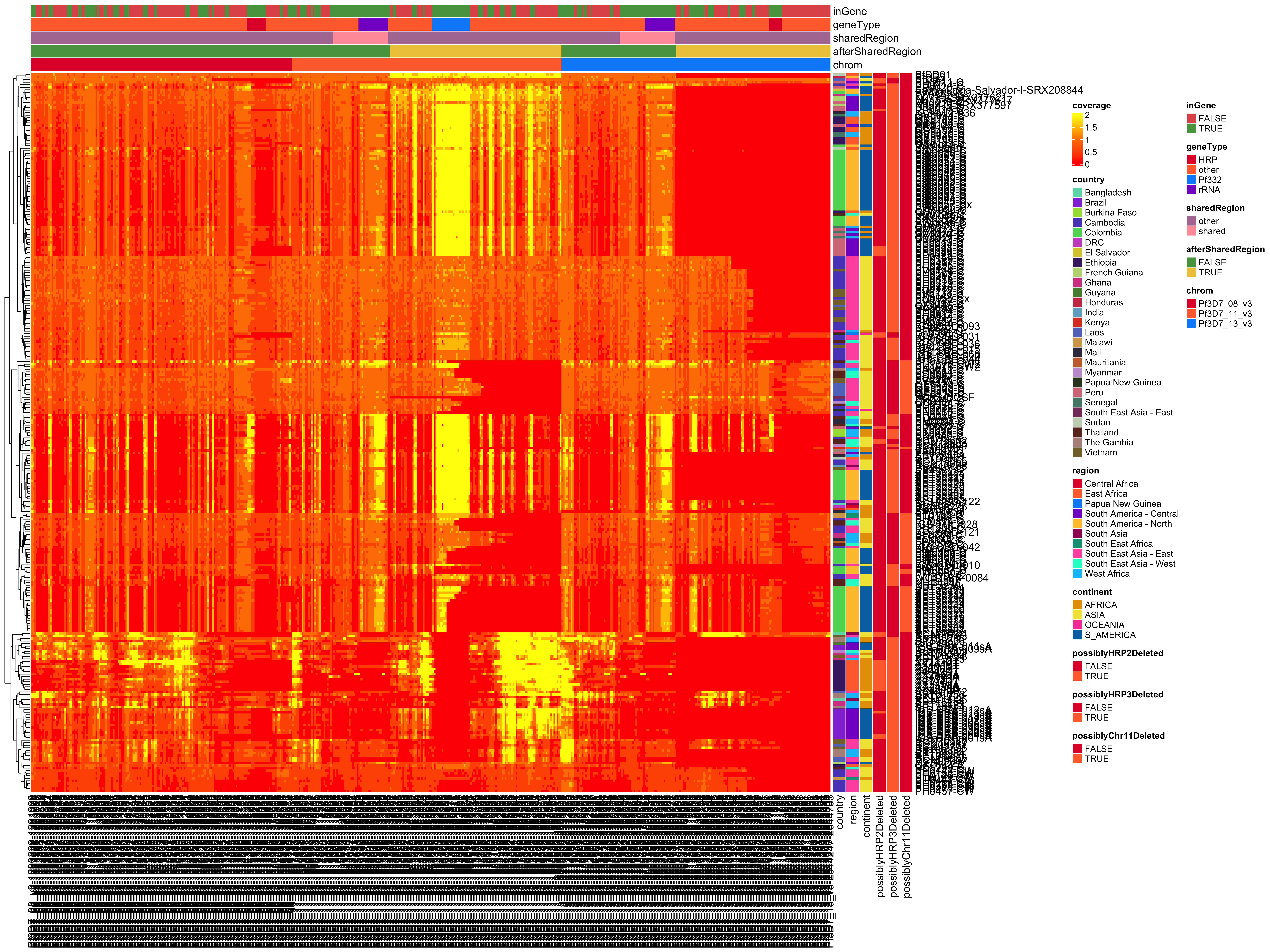
--- title: "Processing Coverage" --- ```{r setup, echo=FALSE, message=FALSE} source("../common.R") ``` ## Read in meta and genome whole coverage and Finding low coverage in HRP gene areas ```{r} = readr:: read_tsv ("../meta/metadata/meta.tab.txt" )= allMeta %>% filter (is.na (site) | site != "LabContaminated" , = "data/finalHRPII_HRPIII_windows_withTuned/popClustering/reports/allBasicInfo.tab.txt.gz" = "initialCoverageDataObjects.Rdata" #file.remove(outputFnp) if (! file.exists (outputFnp) | file.info (inputFnp)$ ctime > file.info (outputFnp)$ ctime){= readr:: read_tsv ("../meta/allCov_summaryStats.tab.txt.gz" )= readr:: read_tsv (inputFnp) %>% filter (sample %fin% allMeta_filt$ sample)= allBasicInfo %>% left_join (cov)= allBasicInfo %>% group_by (sample) %>% summarise (medianCov = median (perBaseCoverage), meanCov = mean (perBaseCoverage)) = allBasicInfo_sampleCoverageStats %>% filter (medianCov > 5 )= allBasicInfo %>% filter (sample %in% allBasicInfo_sampleCoverageStats_highCov$ sample)= allBasicInfo_higCov %>% mutate (marker = ifelse (perBaseCoverage < 1 , 1 , 0 )) %>% group_by (sample, ` #chrom ` ) %>% summarise (lowCov = sum (marker))#these below regions have variable cross mapping in other strains = c ("Pf3D7_13_v3-2852656-2852831" , "Pf3D7_13_v3-2853081-2853381" , "Pf3D7_13_v3-2853978-2854303" , "Pf3D7_13_v3-2855203-2855353" )# filter coverage data to specific regions of interest = allBasicInfo_higCov %>% filter ((` #chrom ` == "Pf3D7_13_v3" & start > 2840426 & start < 2842301 ) | ` #chrom ` == "Pf3D7_11_v3" & start > 1982395 ) | ` #chrom ` == "Pf3D7_08_v3" & start >= 1374235 & start < 1375485 ))= cov %>% filter (medianPerBaseCov_inGenes > 100 )# now process to determine which samples potentially have deleted regions of interest base on coverage = allBasicInfo_higCov_areasOfInterest %>% mutate (perBaseCoverage = ifelse (readTotal <= ifelse (sample %in% cov_highMedCov$ sample, 20 , 10 ) & readTotalUsed == 0 & fullySpanningReads <= 1 , 0 , perBaseCoverage))$ processedMarker = 0 $ processedSuccessMarker = 0 for (row in 2 : (nrow (allBasicInfo_higCov_areasOfInterest) - 1 )){#print(row) if (allBasicInfo_higCov_areasOfInterest$ sample[row] == allBasicInfo_higCov_areasOfInterest$ sample[row + 1 ]& allBasicInfo_higCov_areasOfInterest$ ` #chrom ` [row] == allBasicInfo_higCov_areasOfInterest$ ` #chrom ` [row + 1 ]& allBasicInfo_higCov_areasOfInterest$ perBaseCoverage[row - 1 ] == 0 & allBasicInfo_higCov_areasOfInterest$ perBaseCoverage[row] == 0 & allBasicInfo_higCov_areasOfInterest$ perBaseCoverage[row + 1 ] == 0 # & allBasicInfo_higCov_areasOfInterest$perBaseCoverage[row - 1] < 0.1 # & allBasicInfo_higCov_areasOfInterest$perBaseCoverage[row] < 0.1 # & allBasicInfo_higCov_areasOfInterest$perBaseCoverage[row + 1] <0.1 $ processedMarker[row] = 1 if (allBasicInfo_higCov_areasOfInterest$ sample[row] == allBasicInfo_higCov_areasOfInterest$ sample[row + 1 ]& allBasicInfo_higCov_areasOfInterest$ ` #chrom ` [row] == allBasicInfo_higCov_areasOfInterest$ ` #chrom ` [row + 1 ]& allBasicInfo_higCov_areasOfInterest$ success[row - 1 ] == F& allBasicInfo_higCov_areasOfInterest$ success[row] == F& allBasicInfo_higCov_areasOfInterest$ success[row + 1 ] == F# & allBasicInfo_higCov_areasOfInterest$perBaseCoverage[row - 1] < 0.1 # & allBasicInfo_higCov_areasOfInterest$perBaseCoverage[row] < 0.1 # & allBasicInfo_higCov_areasOfInterest$perBaseCoverage[row + 1] <0.1 $ processedSuccessMarker[row] = 1 save (cov,file = outputFnp)else {load (outputFnp)= allBasicInfo_higCov_areasOfInterest %>% #filter(!is.na(extraField0)) %>% # mutate(marker = ifelse(perBaseCoverage < 0.1, 1, 0)) %>% mutate (marker = ifelse (perBaseCoverage == 0 , 1 , 0 )) %>% mutate (successMarker = ifelse (! success, 1 , 0 )) %>% group_by (sample, ` #chrom ` ) %>% summarise (lowCov = sum (marker),lowCovStrict = sum (processedMarker),lowSuccess = sum (successMarker),lowSuccessStrict = sum (processedSuccessMarker),total = n ()%>% mutate (lowCovPerc = lowCov / total,lowCovStrictPerc = lowCovStrict / (total - 2 ),lowSuccessPerc = lowSuccess / total,lowSuccessStrictPerc = lowSuccessStrict / (total - 2 )# there are several samples which due to high variability in coverage, all of which are sWGA samples or in other artifacts introduced by WGS processes that can't be easily filtered out programatically so # they are removed based on analysis by hand = c (# chr08 hand investigations "P178-D30" , "PD1192-C" , "IGS-BRA-018sA" , "PA0331-CW" , "T367" , "RCN09021" ,"RCN13559" , "RCN09011" , "RCN11871" , "RCN02972" , "SPT35541" , "T508" ,# chr08 hand investigations on finer windows "SPT24525" , "SPT24643" , "SPT24651" , "SPT24541" , "SPT24646" , "SPT24530" , "SPT24607" , "SPT24650" ,# chr13 hand investigations "SPT22213" , "RCN08027" , "RCN12032" , "SPT35322" , "SPT35228" , "SPT35354" ,"RCN02907" , "RCN02953" , "RCN08023" , "PF1157-Cx6" , "IGS-BRA-016sA" , "IGS-BRA-018sA" , "NHP4374" , "PA0809-CW" , "SPT21769" , "SPT35543" , "T498" , "T395" , "RCN02816" , "RCN02821" , "RCN02893" , "RCN03336" , "RCN03536" , "RCN02547" , "SPT21675" , "PA0331-CW" , "RCN14050" ,"SPT16994" ,"SPT38383" ,"SPT40863" ,"SPT38318" ,"SPT15609" ,"SPT38344" ,"SPT34334" ,"SPT38314" ,"SPT34321" ,"SPT17972" ,"SPT19692" ,"SPT12080" ,# chr13 hand investigations on finer windows "RCN13051" , "PR0262-C" , "SPT00902" , "SPT00903" , "SPT00898" , "SPT26258" , "SPT26293" , "SPT26254" ,"PW0099-C" ,"SPT24499" , "RCN13387" , "SPT21927" , "RCN08119" ,"RCN13415" , "RCN13414" , "RCN13413" ,"RCN13391" , "RCN13388" , "RCN11770" , "RCN11771" , "RCN13416" , "SPT21927" , "RCN09061" , "RCN11766" , "RCN13490" , "SPT25144" , "RCN09058" , "RCN09027" , "RCN09064" ,"RCN12064" , "RCN11497" , "RCN02091" , "SPT18468" , "RCN13544" , "RCN11601" , "SPT24457" , "SPT43231" , "RCN02894" , "RCN02146" , "RCN02365" , "RCN02171" ,"SPT15757" , "SPT35260" ,# chr11 hand investigations "SPT43093" , "RCN13306" ,"RCN13447" ,"RCN11811" ,"SPT43196" ,"RCN11158" ,"RCN11805" ,"RCN13308" ,"RCN13309" ,"RCN13269" ,"RCN13285" ,"RCN13267" ,"SPT43280" ,"RCN02046" ,"RCN13296" ,"RCN13323" ,"RCN13310" ,"RCN09041" ,"RCN09043" ,"RCN09042" ,"RCN09071" ,"RCN11693" ,"RCN11696" ,"RCN11687" ,"RCN11644" ,"RCN09157" ,"RCN10396" ,"RCN11167" ,"RCN11223" ,"RCN10306" ,"RCN00127" ,"RCN00266" ,"RCN09029" ,"RCN00129" ,"RCN02982" ,"RCN03040" ,"PM0744-CW" ,"RCN08298" ,"RCN11172" ,"RCN11234" ,"RCN10247" ,"RCN10297" ,"PA0295-CW" ,"PR0006-CW" ,"NHP4224" ,"NHP4850" ,"RCN11636" ,"RCN13441" ,"RCN13324" ,"RCN09074" ,"RCN13437" ,"RCN09073" ,"RCN11494" ,"RCN11611" ,"RCN13515" ,"RCN13523" ,"RCN11787" ,"RCN13532" ,"RCN02848" ,"SPT35212" ,"RCN11616" ,"RCN13336" ,"RCN13421" ,"SPT42050" ,"RCN13420" ,"RCN13481" ,"SPT36128" ,"SPT34434" ,"SPT36696" ,"SPT38311" ,"RCN13417" ,"SPT34254" ,"RCN02341" ,"RCN13442" ,"RCN13911" ,"SPT19293" ,"SPT34315" ,"SPT34330" ,"SPT34327" ,"SPT34335" # further removing SWGA samples and control mixtures = allBasicInfo_higCov_areasOfInterest_covLessThan1Sum %>% filter (! grepl ("^PG" , sample)) %>% filter (! grepl ("^[0-9]S[0-9]-[0-9]C[0-9]+" , sample)) %>% filter (! grepl ("^SenT" , sample)) %>% filter (! grepl ("^80[0-9]+" , sample)) %>% filter (! grepl ("^OHP[0-9]+" , sample)) %>% filter (! grepl ("^ERR" , sample)) %>% filter (! (sample %in% handRemovedSamples)) %>% filter (lowCovPerc >= .666 & lowCovStrictPerc >= 0.33 ) # %>% # filter((lowCovPerc >= .666 & lowCovStrictPerc >= 0.50) | lowSuccessPerc > 0.95) # = allBasicInfo_higCov_areasOfInterest_covLessThan1Sum %>% filter (! grepl ("^PG" , sample)) %>% filter (! grepl ("^[0-9]S[0-9]-[0-9]C[0-9]+" , sample)) %>% filter (! grepl ("^SenT" , sample)) %>% filter (! grepl ("^80[0-9]+" , sample)) %>% filter (! grepl ("^OHP[0-9]+" , sample)) %>% filter (! grepl ("^ERR" , sample)) %>% filter (! (sample %in% handRemovedSamples)) %>% filter (! (lowCovPerc >= .666 & lowCovStrictPerc >= 0.33 ) )# %>% # filter(!((lowCovPerc >= .666 & lowCovStrictPerc >= 0.50) | lowSuccessPerc > 0.95) ) # cat (unique (c (allBasicInfo_higCov_areasOfInterest_covLessThan1Sum_highLowCov_opposite$ sample, $ sample)), file = "samplesCovered.txt" , sep = " \n " )= allBasicInfo_higCov_areasOfInterest_covLessThan1Sum_highLowCov %>% filter (` #chrom ` == "Pf3D7_08_v3" )= allBasicInfo_higCov_areasOfInterest_covLessThan1Sum_highLowCov %>% filter (` #chrom ` == "Pf3D7_13_v3" )= allBasicInfo_higCov_areasOfInterest_covLessThan1Sum_highLowCov %>% filter (` #chrom ` == "Pf3D7_11_v3" )= allBasicInfo %>% filter (sample %fin% allBasicInfo_higCov_areasOfInterest_covLessThan1Sum_highLowCov$ sample) %>% group_by (sample) %>% mutate (perBaseCoverageNorm = perBaseCoverage/ medianPerBaseCov_inGenes)%>% mutate (perBaseCoverageNormRounded = round (perBaseCoverageNorm/ 0.5 )* 0.5 ) %>% mutate (id = paste0 (` #chrom ` , "-" , start, "-" , end)) = c ("Pf3D7_08_v3-1382255-1382505" ,"Pf3D7_08_v3-1382680-1383155" ,"Pf3D7_08_v3-1384030-1384251" ,"Pf3D7_08_v3-1384316-1384663" ,"Pf3D7_08_v3-1384837-1385370" ,#"Pf3D7_08_v3-1385625-1385741", "Pf3D7_08_v3-1385951-1386323" ,"Pf3D7_08_v3-1386518-1386680" ,"Pf3D7_08_v3-1386739-1387414" ,"Pf3D7_08_v3-1387782-1387982" , "Pf3D7_11_v3-1997276-1997425" ,"Pf3D7_11_v3-1997674-1997884" ,"Pf3D7_11_v3-1997944-1998371" ,"Pf3D7_11_v3-1998638-1998759" ,"Pf3D7_11_v3-1998833-1999151" ,"Pf3D7_11_v3-1999245-1999390" ,"Pf3D7_11_v3-1999600-1999713" ,"Pf3D7_11_v3-2000687-2000889" ,"Pf3D7_11_v3-2000941-2001239" ,"Pf3D7_11_v3-2001300-2003328" , "Pf3D7_13_v3-2842251-2842401" ,"Pf3D7_13_v3-2842251-2842451" ,"Pf3D7_13_v3-2842301-2842601" ,"Pf3D7_13_v3-2842476-2842688" ,"Pf3D7_13_v3-2842501-2842651" ,"Pf3D7_13_v3-2842515-2842804" ,"Pf3D7_13_v3-2843108-2843446" ,"Pf3D7_13_v3-2843641-2843863" ,"Pf3D7_13_v3-2843930-2844101" ,"Pf3D7_13_v3-2844247-2844785" # "Pf3D7_13_v3-2842501-2842576","Pf3D7_13_v3-2842501-2842601","Pf3D7_13_v3-2842501-2842651","Pf3D7_13_v3-2842515-2842804","Pf3D7_13_v3-2842576-2842651", # "Pf3D7_13_v3-2842601-2842688","Pf3D7_13_v3-2843108-2843446","Pf3D7_13_v3-2843641-2843863","Pf3D7_13_v3-2843930-2844101","Pf3D7_13_v3-2844247-2844785" # ## ## after careful inspection of the samples, all potential true deletion appear to include the whole chromosome from the point of interest so in order to clean up the samples will force samples ## to have no coverage from the point of interest on to get high quality examples of chromosomal deletions of areas of interest = c ("Pf3D7_08_v3-1375557-1375750" ,"Pf3D7_08_v3-1378006-1378662" ,"Pf3D7_08_v3-1379154-1379915" ,"Pf3D7_08_v3-1380194-1380328" ,"Pf3D7_08_v3-1382255-1382505" ,"Pf3D7_08_v3-1382680-1383155" ,"Pf3D7_08_v3-1384030-1384251" ,"Pf3D7_08_v3-1384316-1384663" ,"Pf3D7_08_v3-1384837-1385370" ,#"Pf3D7_08_v3-1385625-1385741", "Pf3D7_08_v3-1385951-1386323" ,"Pf3D7_08_v3-1386518-1386680" ,"Pf3D7_08_v3-1386739-1387414" ,"Pf3D7_08_v3-1387782-1387982" )= c ("Pf3D7_11_v3-1991347-1992851" ,"Pf3D7_11_v3-1992907-1993669" ,"Pf3D7_11_v3-1993833-1994061" ,"Pf3D7_11_v3-1994139-1994363" ,"Pf3D7_11_v3-1995234-1995394" ,"Pf3D7_11_v3-1995459-1995614" ,"Pf3D7_11_v3-1995678-1996112" ,"Pf3D7_11_v3-1996349-1996650" ,"Pf3D7_11_v3-1996745-1997019" ,"Pf3D7_11_v3-1997095-1997221" ,"Pf3D7_11_v3-1997276-1997425" ,"Pf3D7_11_v3-1997674-1997884" ,"Pf3D7_11_v3-1997944-1998371" ,"Pf3D7_11_v3-1998638-1998759" ,"Pf3D7_11_v3-1998833-1999151" ,"Pf3D7_11_v3-1999245-1999390" ,"Pf3D7_11_v3-1999600-1999713" ,"Pf3D7_11_v3-2000687-2000889" ,"Pf3D7_11_v3-2000941-2001239" ,"Pf3D7_11_v3-2001300-2003328" )= c ("Pf3D7_13_v3-2841776-2841926" ,"Pf3D7_13_v3-2842001-2842301" ,"Pf3D7_13_v3-2842026-2842226" ,"Pf3D7_13_v3-2842038-2842461" ,"Pf3D7_13_v3-2842101-2842251" ,"Pf3D7_13_v3-2842251-2842401" ,"Pf3D7_13_v3-2842251-2842451" ,"Pf3D7_13_v3-2842301-2842601" ,"Pf3D7_13_v3-2842476-2842688" ,"Pf3D7_13_v3-2842501-2842651" ,"Pf3D7_13_v3-2842515-2842804" ,"Pf3D7_13_v3-2843108-2843446" ,"Pf3D7_13_v3-2843641-2843863" ,"Pf3D7_13_v3-2843930-2844101" ,"Pf3D7_13_v3-2844247-2844785" )#### filter ends chr08 -begin = allBasicInfo_highCov_butLowInAreasofInterest %>% filter (` #chrom ` == "Pf3D7_08_v3" ) %>% filter (%in% allBasicInfo_higCov_areasOfInterest_covLessThan1Sum_highLowCov_chrom08$ sample%>% group_by (sample, id) %>% mutate (marker = ifelse (perBaseCoverageNormRounded == 0 , 1 , 0 )) %>% filter (id %in% endRegions_08) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker))= allBasicInfo_highCov_butLowInAreasofInterest_endsNoCovSel_08 %>% filter (noCov == length (endRegions_08))= allBasicInfo_highCov_butLowInAreasofInterest %>% filter (` #chrom ` == "Pf3D7_08_v3" ) %>% filter (%in% allBasicInfo_higCov_areasOfInterest_covLessThan1Sum_highLowCov_chrom08$ sample%>% group_by (sample, id) %>% mutate (marker = ifelse (success, 0 , 1 )) %>% filter (id %in% endRegions_08) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker))= allBasicInfo_highCov_butLowInAreasofInterest_endsNoSuccessSel_08 %>% filter (noCov == length (endRegions_08))= intersect (sort ($ samplesort ($ sample#### filter ends chr08 -end #### filter ends chr11 -begin = allBasicInfo_highCov_butLowInAreasofInterest %>% filter (` #chrom ` == "Pf3D7_11_v3" ) %>% filter (%in% allBasicInfo_higCov_areasOfInterest_covLessThan1Sum_highLowCov_chrom11$ sample%>% group_by (sample, id) %>% mutate (marker = ifelse (perBaseCoverageNormRounded == 0 , 1 , 0 )) %>% filter (id %in% endRegions_11) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker))= allBasicInfo_highCov_butLowInAreasofInterest_endsNoCovSel_11 %>% filter (noCov == length (endRegions_11))= allBasicInfo_highCov_butLowInAreasofInterest %>% filter (` #chrom ` == "Pf3D7_11_v3" ) %>% filter (%in% allBasicInfo_higCov_areasOfInterest_covLessThan1Sum_highLowCov_chrom11$ sample%>% group_by (sample, id) %>% mutate (marker = ifelse (success, 0 , 1 )) %>% filter (id %in% endRegions_11) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker))= allBasicInfo_highCov_butLowInAreasofInterest_endsNoSuccessSel_11 %>% filter (noCov == length (endRegions_11))= intersect (sort ($ samplesort ($ sample#### filter ends chr11 -end #### filter ends chr13 -begin = allBasicInfo_highCov_butLowInAreasofInterest %>% filter (` #chrom ` == "Pf3D7_13_v3" ) %>% filter (%in% allBasicInfo_higCov_areasOfInterest_covLessThan1Sum_highLowCov_chrom13$ sample%>% group_by (sample, id) %>% mutate (marker = ifelse (perBaseCoverageNormRounded == 0 , 1 , 0 )) %>% filter (id %in% endRegions_13) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker))= allBasicInfo_highCov_butLowInAreasofInterest_endsNoCovSel_13 %>% filter (noCov == length (endRegions_13))= allBasicInfo_highCov_butLowInAreasofInterest %>% filter (` #chrom ` == "Pf3D7_13_v3" ) %>% filter (%in% allBasicInfo_higCov_areasOfInterest_covLessThan1Sum_highLowCov_chrom13$ sample%>% group_by (sample, id) %>% mutate (marker = ifelse (success, 0 , 1 )) %>% filter (id %in% endRegions_13) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker))= allBasicInfo_highCov_butLowInAreasofInterest_endsNoSuccessSel_13 %>% filter (noCov == length (endRegions_13))= intersect (sort ($ samplesort ($ sample#### filter ends chr13 -end = allBasicInfo_highCov_butLowInAreasofInterest %>% group_by (sample, id) %>% mutate (marker = ifelse (perBaseCoverageNormRounded == 0 , 1 , 0 )) %>% filter (id %in% endRegions) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker))= allBasicInfo_highCov_butLowInAreasofInterest_endsNoCovSel %>% filter (noCov == 10 )= allBasicInfo_highCov_butLowInAreasofInterest %>% group_by (sample, id) %>% mutate (marker = ifelse (success, 0 , 1 )) %>% filter (id %in% endRegions) %>% group_by (sample, ` #chrom ` ) %>% summarise (noCov = sum (marker))= allBasicInfo_highCov_butLowInAreasofInterest_endsNoSuccessSel %>% filter (noCov == 10 )= allBasicInfo_highCov_butLowInAreasofInterest %>% # filter(sample %in% allBasicInfo_highCov_butLowInAreasofInterest_endsNoSuccessSel_filt$sample & # sample %in% allBasicInfo_highCov_butLowInAreasofInterest_endsNoCovSel_filt$sample) %>% filter (%in% c (%>% select (sample, id, perBaseCoverageNorm) %>% spread (id, perBaseCoverageNorm)# hold = readr::read_tsv( # "/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/hrps/windowSelectionSurroundingHRPs/Windows_Surrounding_HRP2_3_deletions/windowAnalysis/allMeta_HRP2_HRP3_deletionCalls.tab.txt" # ) = as.matrix (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp[, 2 : ncol (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp)])rownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat) = allBasicInfo_highCov_butLowInAreasofInterest_cov_sp$ sample#cap it at 2 > 2 ] = 2 = gsub ("-.*" , "" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat))# get the regions = allBasicInfo %>% filter (.$ sample[1 ] == sample) %>% select (1 : 6 , extraField0) %>% unique ()%>% mutate (genomicID = paste0 (` #chrom ` , "-" , start, "-" , end)) # rename them based on importance = regions%>% mutate (id = paste0 (` #chrom ` , "-" , start, "-" , end)) %>% arrange (id) %>% mutate (inGene = ! is.na (extraField0)) %>% mutate (geneType = ifelse (grepl ("histidine-rich protein II" , extraField0), "HRP" , "other" ) )%>% mutate (geneType = ifelse (grepl ("ribosomal RNA" , extraField0), "rRNA" , geneType ) )%>% mutate (geneType = ifelse (grepl ("332" , extraField0), "Pf332" , geneType ) ) %>% mutate (sharedRegion = ifelse ((` #chrom ` == "Pf3D7_11_v3" & >= 1918028 & <= 1933288 ) | ` #chrom ` == "Pf3D7_13_v3" & >= 2792021 & <= 2807295 ,"shared" ,"other" %>% mutate (afterSharedRegion = (` #chrom ` == "Pf3D7_11_v3" & >= 1933288 ) | ` #chrom ` == "Pf3D7_13_v3" & >= 2807295 )) %>% mutate (chrom = ` #chrom ` )%>% mutate (description = gsub (".*description=" , "" , extraField0)) %>% mutate (description = ifelse ("[extraField0=NA]" == extraField0, NA , description)) %>% mutate (description = gsub ("]" , "" , fixed = T, description))= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat), regions$ id),]= round (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat/ 0.5 )* 0.5 = readr:: read_tsv ("../meta/metadata/meta.tab.txt" )= allMeta %>% filter ( sample %in% samples_chr13_noCovNoSucEnds | %in% samples_chr08_noCovNoSucEnds | %in% samples_chr11_noCovNoSucEnds ) $ possiblyHRP2Deleted = allMeta_filt$ sample %in% samples_chr08_noCovNoSucEnds$ possiblyHRP3Deleted = allMeta_filt$ sample %in% samples_chr13_noCovNoSucEnds$ possiblyChr11Deleted = allMeta_filt$ sample %in% samples_chr11_noCovNoSucEnds= allMeta_filt[match (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp$ sample, allMeta_filt$ sample), ]``` ## Heatmap of coverage ```{r} = 10 library (circlize)= colorRamp2 (c (0 , 1 , 2 ), c (heat.colors (3 )))= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_roundrownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_noLabs) = NULL colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_noLabs) = NULL = metaSelected$ BiologicalSample$ site != "LabIsolate" | is.na (metaSelected$ site)] = "" rownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_noLabs) = RowLabs= sort (unique (c (metaSelected$ country, metaSelected$ region, metaSelected$ secondaryRegion)))= scheme$ hex (length (regionNames))names (regionColors) = regionNames= regions[,c ("inGene" , "geneType" , "sharedRegion" , "afterSharedRegion" , "chrom" )] %>% as.data.frame ()= createColorListFromDf (topAnnoDf) "inGene" ]] = c ("TRUE" = tableau_color_pal ()(8 )[5 ], "FALSE" = paste0 (tableau_color_pal ()(8 )[3 ],"FF" ))"sharedRegion" ]] = c ("other" = paste0 (tableau_color_pal ()(8 )[7 ], "FF" ), "shared" = tableau_color_pal ()(8 )[8 ])"afterSharedRegion" ]] = c ("TRUE" = tableau_color_pal ()(8 )[6 ], "FALSE" = paste0 (tableau_color_pal ()(8 )[5 ],"FF" ))= metaSelected[,c ("country" , "region" , "secondaryRegion" ,"possiblyHRP2Deleted" , "possiblyHRP3Deleted" , "possiblyChr11Deleted" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()= createColorListFromDf (rowAnnoDf) # rowAnnoColors[["continent"]] = c("AFRICA" = tableau_color_pal()(8)[1], # "ASIA" = tableau_color_pal()(8)[7], # "OCEANIA" = tableau_color_pal()(8)[2], # "S_AMERICA" = tableau_color_pal()(8)[3]) "continent" ]] = c ("AFRICA" = "#E69F00" , "ASIA" = "#F0E442" , "OCEANIA" = "#F748A5" , "S_AMERICA" = "#0072B2" )= rowAnnotation (df = rowAnnoDf, col = rowAnnoColors)= HeatmapAnnotation (df = topAnnoDf, col = topAnnoColors)= rowAnnotation (df = rowAnnoDf, col = rowAnnoColors)= Heatmap (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round, cluster_columns = F, col = col_fun,name = "coverage" ,top_annotation = topAnno, right_annotation = sideAnno)``` ```{r} #| fig-column: screen #| fig-height: 15 #| fig-width: 20 print (allCoverageHeatMap)``` ```{r} pdf ("test_allHeatmap_initial_windows.pdf" , useDingbats = F, width = 20 , height = 50 )print (allCoverageHeatMap)dev.off ()``` ## by chrom ### All ```{r} = readr:: read_tsv ("../meta/metadata/meta.tab.txt" )$ possiblyHRP2Deleted = allMeta$ sample %in% samples_chr08_noCovNoSucEnds$ possiblyHRP3Deleted = allMeta$ sample %in% samples_chr13_noCovNoSucEnds$ possiblyChr11Deleted = allMeta$ sample %in% samples_chr11_noCovNoSucEnds= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_roundlibrary (dendsort)= dendsort (hclust (dist (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)))= allMeta[match (rownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod), allMeta$ sample), ]= metaSelectedForSorting %>% select (sample, possiblyHRP2Deleted, possiblyHRP3Deleted, possiblyChr11Deleted)= as.matrix (metaSelectedForSorting_sel[,2 : ncol (metaSelectedForSorting_sel)])rownames (metaSelectedForSorting_sel_mat) = metaSelectedForSorting_sel$ sample= hclust (dist (metaSelectedForSorting_sel_mat))= metaSelectedForSorting %>% left_join (tibble (sample = rownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)[row_dend$ order], coverageOrder = 1 : length (row_dend$ order)) ) %>% left_join (tibble (sample = rownames (metaSelectedForSorting_sel_mat)[hclust (dist (metaSelectedForSorting_sel_mat))$ order], deletionOrder = 1 : length (hclust (dist (metaSelectedForSorting_sel_mat))$ order)) )= metaSelectedForSorting %>% arrange (desc (possiblyHRP2Deleted), desc (possiblyHRP3Deleted), desc (possiblyChr11Deleted), coverageOrder)= metaSelectedForSorting %>% group_by (sample) %>% mutate (forSortingOld = paste0 (possiblyHRP2Deleted, "-" , possiblyHRP3Deleted, "-" , possiblyChr11Deleted, "-" , stringr:: str_pad (coverageOrder, side = "left" , pad = "0" , width = nchar (max (metaSelectedForSorting$ coverageOrder))))) %>% mutate (forSorting = paste0 (possiblyHRP2Deleted, "-" , possiblyHRP3Deleted, "-" , possiblyChr11Deleted)) %>% ungroup () %>% mutate (forSorting = factor (forSorting, levels = c ("TRUE-FALSE-FALSE" ,"TRUE-TRUE-FALSE" , "FALSE-TRUE-FALSE" , "FALSE-TRUE-TRUE" , "FALSE-FALSE-TRUE" ))) %>% arrange (forSorting, coverageOrder)= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[match (metaSelectedForSorting$ sample,rownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)),]= allMeta[match (rownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod), allMeta$ sample), ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_08_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_11_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_13_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= metaSelected[,c ("country" , "region" , "secondaryRegion" ,"possiblyHRP2Deleted" , "possiblyHRP3Deleted" , "possiblyChr11Deleted" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()= createColorListFromDf (rowAnnoDf) "continent" ]] = c ("AFRICA" = tableau_color_pal ()(8 )[1 ], "ASIA" = tableau_color_pal ()(8 )[7 ], "OCEANIA" = tableau_color_pal ()(8 )[2 ], "S_AMERICA" = tableau_color_pal ()(8 )[3 ])= rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),simple_anno_size = unit (1.5 , "cm" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )gap = unit (0.1 , "cm" )= createColorListFromDf (regions %>% select (c ("inGene" , "geneType" , "sharedRegion" , "afterSharedRegion" , "chrom" , "description" ))) "inGene" ]] = c ("TRUE" = tableau_color_pal ()(8 )[5 ], "FALSE" = paste0 (tableau_color_pal ()(8 )[3 ],"FF" ))"sharedRegion" ]] = c ("other" = paste0 (tableau_color_pal ()(8 )[7 ], "FF" ), "shared" = tableau_color_pal ()(8 )[8 ])"afterSharedRegion" ]] = c ("TRUE" = tableau_color_pal ()(8 )[6 ], "FALSE" = paste0 (tableau_color_pal ()(8 )[5 ],"FF" ))= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08), regions$ genomicID),]= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11), regions$ genomicID),]= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13), regions$ genomicID),]= regions_chr08 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr08_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = T,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )= regions_chr11 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr11_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = F,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )= regions_chr13 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr13_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = F,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08_noColLabs) = NULL = Heatmap (cluster_columns = F,# cluster_rows = row_dend, cluster_rows = F, col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr08,left_annotation = sideAnno,column_title = "Chr08" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11_noColLabs) = NULL = Heatmap (cluster_columns = F,# cluster_rows = row_dend, cluster_rows = F, col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr11,#left_annotation = sideAnno, column_title = "Chr11" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13_noColLabs) = NULL = Heatmap (cluster_columns = F,# cluster_rows = row_dend, cluster_rows = F, col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr13,#left_annotation = sideAnno, column_title = "Chr13" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )``` ```{r} = heatmap_08 + heatmap_11 + heatmap_13``` ```{r} cairo_pdf ("test_allHeatmap_initial_windows_byChrom.pdf" , width = 20 , height = 50 )draw (heatmaps_by_chrom, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "bottom" , annotation_legend_side = "bottom" , ht_gap = unit (2 , "cm" ))dev.off ()``` ### hrp2 deleted ```{r} = readr:: read_tsv ("/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/allSRAData/update_2023_01/Pf7_samples.txt" )= readr:: read_tsv ("../meta/metadata/meta.tab.txt" ) %>% left_join (pf7meta %>% select (Sample, ` Sample type ` ) %>% rename (sample = Sample, seqType = ` Sample type ` ))$ possiblyHRP2Deleted = allMeta$ sample %in% samples_chr08_noCovNoSucEnds$ possiblyHRP3Deleted = allMeta$ sample %in% samples_chr13_noCovNoSucEnds$ possiblyChr11Deleted = allMeta$ sample %in% samples_chr11_noCovNoSucEnds= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round= match (samples_chr08_noCovNoSucEnds, rownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod))= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[selectedSamples[! is.na (selectedSamples)], ]= allMeta[match (rownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod), allMeta$ sample), ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_08_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_11_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_13_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= metaSelected[,c ("country" , "region" , "secondaryRegion" ,"possiblyHRP2Deleted" , "possiblyHRP3Deleted" , "possiblyChr11Deleted" , "seqType" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()= createColorListFromDf (rowAnnoDf) "continent" ]] = c ("AFRICA" = tableau_color_pal ()(8 )[1 ], "ASIA" = tableau_color_pal ()(8 )[7 ], "OCEANIA" = tableau_color_pal ()(8 )[2 ], "S_AMERICA" = tableau_color_pal ()(8 )[3 ])= rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),simple_anno_size = unit (1.5 , "cm" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )gap = unit (0.1 , "cm" )= createColorListFromDf (regions %>% select (c ("inGene" , "geneType" , "sharedRegion" , "afterSharedRegion" , "chrom" , "description" ))) "inGene" ]] = c ("TRUE" = tableau_color_pal ()(8 )[5 ], "FALSE" = paste0 (tableau_color_pal ()(8 )[3 ],"FF" ))"sharedRegion" ]] = c ("other" = paste0 (tableau_color_pal ()(8 )[7 ], "FF" ), "shared" = tableau_color_pal ()(8 )[8 ])"afterSharedRegion" ]] = c ("TRUE" = tableau_color_pal ()(8 )[6 ], "FALSE" = paste0 (tableau_color_pal ()(8 )[5 ],"FF" ))= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08), regions$ genomicID),]= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11), regions$ genomicID),]= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13), regions$ genomicID),]= regions_chr08 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr08_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = T,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )= regions_chr11 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr11_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = F,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )= regions_chr13 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr13_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = F,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )library (dendsort)= dendsort (hclust (dist (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)))= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08_noColLabs) = NULL = Heatmap (cluster_columns = F,cluster_rows = row_dend,col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr08,left_annotation = sideAnno,column_title = "Chr08" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11_noColLabs) = NULL = Heatmap (cluster_columns = F,cluster_rows = row_dend,col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr11,#left_annotation = sideAnno, column_title = "Chr11" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13_noColLabs) = NULL = Heatmap (cluster_columns = F,cluster_rows = row_dend,col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr13,#left_annotation = sideAnno, column_title = "Chr13" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )``` ```{r} = heatmap_08 + heatmap_11 + heatmap_13``` ```{r} cairo_pdf ("test_allHeatmap_initial_windows_byChrom_hrp2_del.pdf" , width = 20 , height = 50 )draw (heatmaps_by_chrom, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "bottom" , annotation_legend_side = "bottom" , ht_gap = unit (2 , "cm" ))dev.off ()``` ### hrp3 deleted ```{r} = readr:: read_tsv ("/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/allSRAData/update_2023_01/Pf7_samples.txt" )= readr:: read_tsv ("../meta/metadata/meta.tab.txt" ) %>% left_join (pf7meta %>% select (Sample, ` Sample type ` ) %>% rename (sample = Sample, seqType = ` Sample type ` ))$ possiblyHRP2Deleted = allMeta$ sample %in% samples_chr08_noCovNoSucEnds$ possiblyHRP3Deleted = allMeta$ sample %in% samples_chr13_noCovNoSucEnds$ possiblyChr11Deleted = allMeta$ sample %in% samples_chr11_noCovNoSucEnds= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round= match (samples_chr13_noCovNoSucEnds, rownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod))= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[selectedSamples[! is.na (selectedSamples)], ]= allMeta[match (rownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod), allMeta$ sample), ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_08_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_11_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_13_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= metaSelected[,c ("country" , "region" , "secondaryRegion" ,"possiblyHRP2Deleted" , "possiblyHRP3Deleted" , "possiblyChr11Deleted" , "seqType" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()= createColorListFromDf (rowAnnoDf) "continent" ]] = c ("AFRICA" = tableau_color_pal ()(8 )[1 ], "ASIA" = tableau_color_pal ()(8 )[7 ], "OCEANIA" = tableau_color_pal ()(8 )[2 ], "S_AMERICA" = tableau_color_pal ()(8 )[3 ])= rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),simple_anno_size = unit (1.5 , "cm" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )gap = unit (0.1 , "cm" )= createColorListFromDf (regions %>% select (c ("inGene" , "geneType" , "sharedRegion" , "afterSharedRegion" , "chrom" , "description" ))) "inGene" ]] = c ("TRUE" = tableau_color_pal ()(8 )[5 ], "FALSE" = paste0 (tableau_color_pal ()(8 )[3 ],"FF" ))"sharedRegion" ]] = c ("other" = paste0 (tableau_color_pal ()(8 )[7 ], "FF" ), "shared" = tableau_color_pal ()(8 )[8 ])"afterSharedRegion" ]] = c ("TRUE" = tableau_color_pal ()(8 )[6 ], "FALSE" = paste0 (tableau_color_pal ()(8 )[5 ],"FF" ))= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08), regions$ genomicID),]= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11), regions$ genomicID),]= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13), regions$ genomicID),]= regions_chr08 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr08_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = T,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )= regions_chr11 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr11_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = F,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )= regions_chr13 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr13_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = F,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )library (dendsort)= dendsort (hclust (dist (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)))= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08_noColLabs) = NULL = Heatmap (cluster_columns = F,cluster_rows = row_dend,col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr08,left_annotation = sideAnno,column_title = "Chr08" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11_noColLabs) = NULL = Heatmap (cluster_columns = F,cluster_rows = row_dend,col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr11,#left_annotation = sideAnno, column_title = "Chr11" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13_noColLabs) = NULL = Heatmap (cluster_columns = F,cluster_rows = row_dend,col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr13,#left_annotation = sideAnno, column_title = "Chr13" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )``` ```{r} = heatmap_08 + heatmap_11 + heatmap_13``` ```{r} cairo_pdf ("test_allHeatmap_initial_windows_byChrom_hrp3_del.pdf" , width = 20 , height = 50 )draw (heatmaps_by_chrom, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "bottom" , annotation_legend_side = "bottom" , ht_gap = unit (2 , "cm" ))dev.off ()``` ### chr11 deleted ```{r} = readr:: read_tsv ("/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/allSRAData/update_2023_01/Pf7_samples.txt" )= readr:: read_tsv ("../meta/metadata/meta.tab.txt" ) %>% left_join (pf7meta %>% select (Sample, ` Sample type ` ) %>% rename (sample = Sample, seqType = ` Sample type ` ))$ possiblyHRP2Deleted = allMeta$ sample %in% samples_chr08_noCovNoSucEnds$ possiblyHRP3Deleted = allMeta$ sample %in% samples_chr13_noCovNoSucEnds$ possiblyChr11Deleted = allMeta$ sample %in% samples_chr11_noCovNoSucEnds= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round= match (rownames (= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[selectedSamples[! is.na (selectedSamples)], ]= allMeta[match (rownames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod), allMeta$ sample), ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_08_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_11_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod[,grepl ("_13_" , colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)) ]= metaSelected[,c ("country" , "region" , "secondaryRegion" ,"possiblyHRP2Deleted" , "possiblyHRP3Deleted" , "possiblyChr11Deleted" , "seqType" )] %>% rename (continent = secondaryRegion) %>% as.data.frame ()= createColorListFromDf (rowAnnoDf) "continent" ]] = c ("AFRICA" = tableau_color_pal ()(8 )[1 ], "ASIA" = tableau_color_pal ()(8 )[7 ], "OCEANIA" = tableau_color_pal ()(8 )[2 ], "S_AMERICA" = tableau_color_pal ()(8 )[3 ])= rowAnnotation (df = rowAnnoDf,col = rowAnnoColors,gp = gpar (col = "grey10" ),simple_anno_size = unit (1.5 , "cm" ),annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )gap = unit (0.1 , "cm" )= createColorListFromDf (regions %>% select (c ("inGene" , "geneType" , "sharedRegion" , "afterSharedRegion" , "chrom" , "description" ))) "inGene" ]] = c ("TRUE" = tableau_color_pal ()(8 )[5 ], "FALSE" = paste0 (tableau_color_pal ()(8 )[3 ],"FF" ))"sharedRegion" ]] = c ("other" = paste0 (tableau_color_pal ()(8 )[7 ], "FF" ), "shared" = tableau_color_pal ()(8 )[8 ])"afterSharedRegion" ]] = c ("TRUE" = tableau_color_pal ()(8 )[6 ], "FALSE" = paste0 (tableau_color_pal ()(8 )[5 ],"FF" ))= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08), regions$ genomicID),]= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11), regions$ genomicID),]= regions[match (colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13), regions$ genomicID),]= regions_chr08 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr08_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = T,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )= regions_chr11 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr11_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = F,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )= regions_chr13 %>% select (inGene, geneType, sharedRegion, afterSharedRegion, chrom, description) %>% as.data.frame ()= HeatmapAnnotation (df = regions_chr13_df,col = regions_cols,annotation_name_gp = gpar (fontsize = annotationTextSize),annotation_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize, fontface = "bold" )annotation_name_side = "left" ,show_annotation_name = F,na_col = "#FFFFFF00" ,gap = unit (0.1 , "cm" )library (dendsort)= dendsort (hclust (dist (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_mod)))= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr08_noColLabs) = NULL = Heatmap (cluster_columns = F,cluster_rows = row_dend,col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr08,left_annotation = sideAnno,column_title = "Chr08" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr11_noColLabs) = NULL = Heatmap (cluster_columns = F,cluster_rows = row_dend,col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr11,#left_annotation = sideAnno, column_title = "Chr11" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )= allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13colnames (allBasicInfo_highCov_butLowInAreasofInterest_cov_sp_mat_round_chr13_noColLabs) = NULL = Heatmap (cluster_columns = F,cluster_rows = row_dend,col = col_fun,name = "coverage" ,row_dend_width = unit (5 , "cm" ),column_dend_height = unit (5 , "cm" ),heatmap_legend_param = list (labels_gp = gpar (fontsize = annotationTextSize),title_gp = gpar (fontsize = annotationTextSize,fontface = "bold" ,title = "coverage" ,at = c (0 , 0.5 , 1 , 1.5 , 2 ),labels = c ("0" , "0.5x" , "1.0x" , "1.5x" , ">=2x" )top_annotation = topAnno_chr13,#left_annotation = sideAnno, column_title = "Chr13" ,column_title_gp = gpar (fontsize = 30 , fontface = "bold" )``` ```{r} = heatmap_08 + heatmap_11 + heatmap_13``` ```{r} cairo_pdf ("test_allHeatmap_initial_windows_byChrom_chr11_del.pdf" , width = 20 , height = 20 )draw (heatmaps_by_chrom, background = "transparent" , merge_legend = TRUE , heatmap_legend_side = "bottom" , annotation_legend_side = "bottom" , ht_gap = unit (2 , "cm" ))dev.off ()``` # meta data counts ```{r} = pf7meta %>% filter (` Sample type ` == "sWGA" )= readr:: read_tsv ("../meta/metadata/meta.tab.txt" )= allMeta %>% filter ( sample %in% samples_chr13_noCovNoSucEnds | %in% samples_chr08_noCovNoSucEnds | %in% samples_chr11_noCovNoSucEnds ) $ possiblyHRP2Deleted = allMeta_filt$ sample %in% samples_chr08_noCovNoSucEnds$ possiblyHRP3Deleted = allMeta_filt$ sample %in% samples_chr13_noCovNoSucEnds$ possiblyChr11Deleted = allMeta_filt$ sample %in% samples_chr11_noCovNoSucEndscreate_dt (allMeta_filt)write_tsv (allMeta_filt,"initial_allMeta_HRP2_HRP3_deletionCalls.tab.txt" )= allMeta_filt %>% group_by (country, possiblyHRP2Deleted, possiblyHRP3Deleted) %>% count ()write_tsv (allMeta_filt_sum,"initial_allMeta_HRP2_HRP3_metaSum_deletionCalls.tab.txt" )create_dt (allMeta_filt_sum)``` ```{r, echo = FALSE, message=FALSE, warning=F, eval=FALSE} #| results: hide pf7meta = readr::read_tsv("/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/allSRAData/update_2023_01/Pf7_samples.txt")%>% filter(!is.na(ENA)) pf7meta_calls = readr::read_tsv("/Users/nick/Dropbox (Personal)/ownCloud/documents/plasmodium/falciparum/pf7/Pf7_inferred_resistance_status_classification_allMeta.tsv") %>% filter(!is.na(ENA)) pf7meta_calls_hrp2_deleted = pf7meta_calls %>% filter(HRP2 == "del") pf7meta_calls_hrp3_deleted = pf7meta_calls %>% filter(HRP3 == "del") pf7meta_calls_hrp2_deleted_filt = pf7meta_calls_hrp2_deleted %>% filter(Sample %!in% samples_chr08_noCovNoSucEnds) pf7meta_calls_hrp3_deleted_filt = pf7meta_calls_hrp3_deleted %>% filter(Sample %!in% samples_chr13_noCovNoSucEnds) temp = pf7meta %>% filter(Sample %in% pf7meta_calls_hrp2_deleted_filt$Sample | Sample %in% pf7meta_calls_hrp3_deleted_filt$Sample) currentSamplesToInvestigate = temp$Sample currentSamplesToInvestigate = (hold %>% filter(sample %!in% allMeta_filt$sample))$sample allBasicInfo_highCov_butLowInAreasofInterest_mod = allBasicInfo %>% filter( sample %fin% currentSamplesToInvestigate ) %>% group_by(sample) %>% mutate(perBaseCoverageNorm = perBaseCoverage / medianPerBaseCov_inGenes) %>% mutate(perBaseCoverageNormRounded = round(perBaseCoverageNorm / 0.5) * 0.5) %>% mutate(id = paste0(`#chrom`, "-", start, "-", end)) allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp = allBasicInfo_highCov_butLowInAreasofInterest_mod %>% select(sample, id, perBaseCoverageNorm) %>% spread(id, perBaseCoverageNorm) allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat = as.matrix(allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp[,2:ncol(allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp)]) rownames(allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat) = allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp$sample #cap it at 2 allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat[allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat>2] =2 chroms = gsub("-.*", "", colnames(allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat)) # get the regions regions = allBasicInfo %>% filter(.$sample[1] == sample) %>% select(1:6, extraField0) %>% unique() # rename them based on importance regions = regions%>% mutate(id = paste0(`#chrom`, "-", start, "-", end)) %>% arrange(id) %>% mutate(inGene = !is.na(extraField0)) %>% mutate(geneType = ifelse(grepl("histidine-rich protein II", extraField0), "HRP", "other" ) )%>% mutate(geneType = ifelse(grepl("ribosomal RNA", extraField0), "rRNA", geneType ) )%>% mutate(geneType = ifelse(grepl("332", extraField0), "Pf332", geneType ) ) %>% mutate(sharedRegion = ifelse((`#chrom` == "Pf3D7_11_v3" & start >= 1918028 & end <= 1933288) | `#chrom` == "Pf3D7_13_v3" & start >= 2792021 & end <= 2807295, "shared", "other" )) %>% mutate(afterSharedRegion = (`#chrom` == "Pf3D7_11_v3" & start >= 1933288) | (`#chrom` == "Pf3D7_13_v3" & start >= 2807295)) %>% mutate(chrom = `#chrom`) regions = regions[match(colnames(allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat), regions$id),] allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat_round = round(allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat/ 0.5)*0.5 metaSelected = allMeta[match(allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp$sample, allMeta$sample), ] library(circlize) col_fun = colorRamp2(c(0, 1, 2), c(heat.colors(3))) allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat_round_noLabs = allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat_round rownames(allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat_round_noLabs) = NULL colnames(allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat_round_noLabs) = NULL RowLabs = metaSelected$BiologicalSample RowLabs[metaSelected$site != "LabIsolate" | is.na(metaSelected$site)] = "" rownames(allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat_round_noLabs) = RowLabs regionNames = sort(unique(c(metaSelected$country, metaSelected$region, metaSelected$secondaryRegion))) regionColors = scheme$hex(length(regionNames)) names(regionColors) = regionNames topAnnoDf = regions[,c("inGene", "geneType", "sharedRegion", "afterSharedRegion", "chrom")] %>% as.data.frame() topAnnoColors = createColorListFromDf(topAnnoDf) topAnnoColors[["inGene"]] = c("TRUE" = tableau_color_pal()(8)[5], "FALSE" = paste0(tableau_color_pal()(8)[3],"FF")) topAnnoColors[["sharedRegion"]] = c("other" = paste0(tableau_color_pal()(8)[7], "FF"), "shared" = tableau_color_pal()(8)[8]) topAnnoColors[["afterSharedRegion"]] = c("TRUE" = tableau_color_pal()(8)[6], "FALSE" = paste0(tableau_color_pal()(8)[5],"FF")) rowAnnoDf = metaSelected[,c("country", "region", "secondaryRegion")] %>% rename(continent = secondaryRegion) %>% as.data.frame() rowAnnoColors = createColorListFromDf(rowAnnoDf) rowAnnoColors[["continent"]] = c("AFRICA" = tableau_color_pal()(8)[1], "ASIA" = tableau_color_pal()(8)[7], "OCEANIA" = tableau_color_pal()(8)[2], "S_AMERICA" = tableau_color_pal()(8)[3]) topAnno = HeatmapAnnotation(df = topAnnoDf, col = topAnnoColors) sideAnno = rowAnnotation(df = rowAnnoDf, col = rowAnnoColors) allCoverageHeatMap = Heatmap(allBasicInfo_highCov_butLowInAreasofInterest_mod_cov_sp_mat_round, cluster_columns = F, col = col_fun, name = "coverage", top_annotation = topAnno, right_annotation = sideAnno) cairo_pdf("investigate_allHeatmap_initial_windows.pdf", width = 20, height = 20) print(allCoverageHeatMap) dev.off() ```