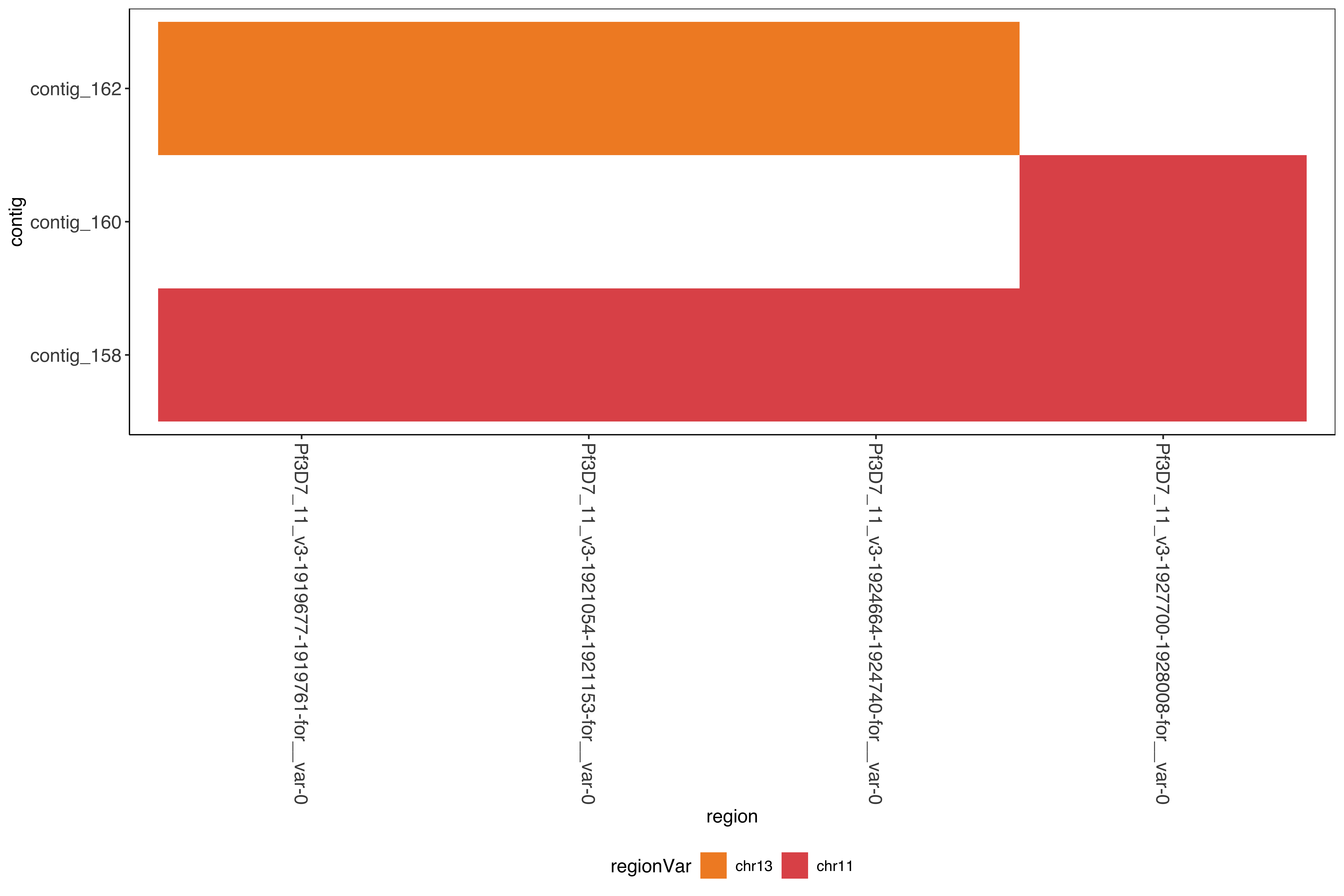
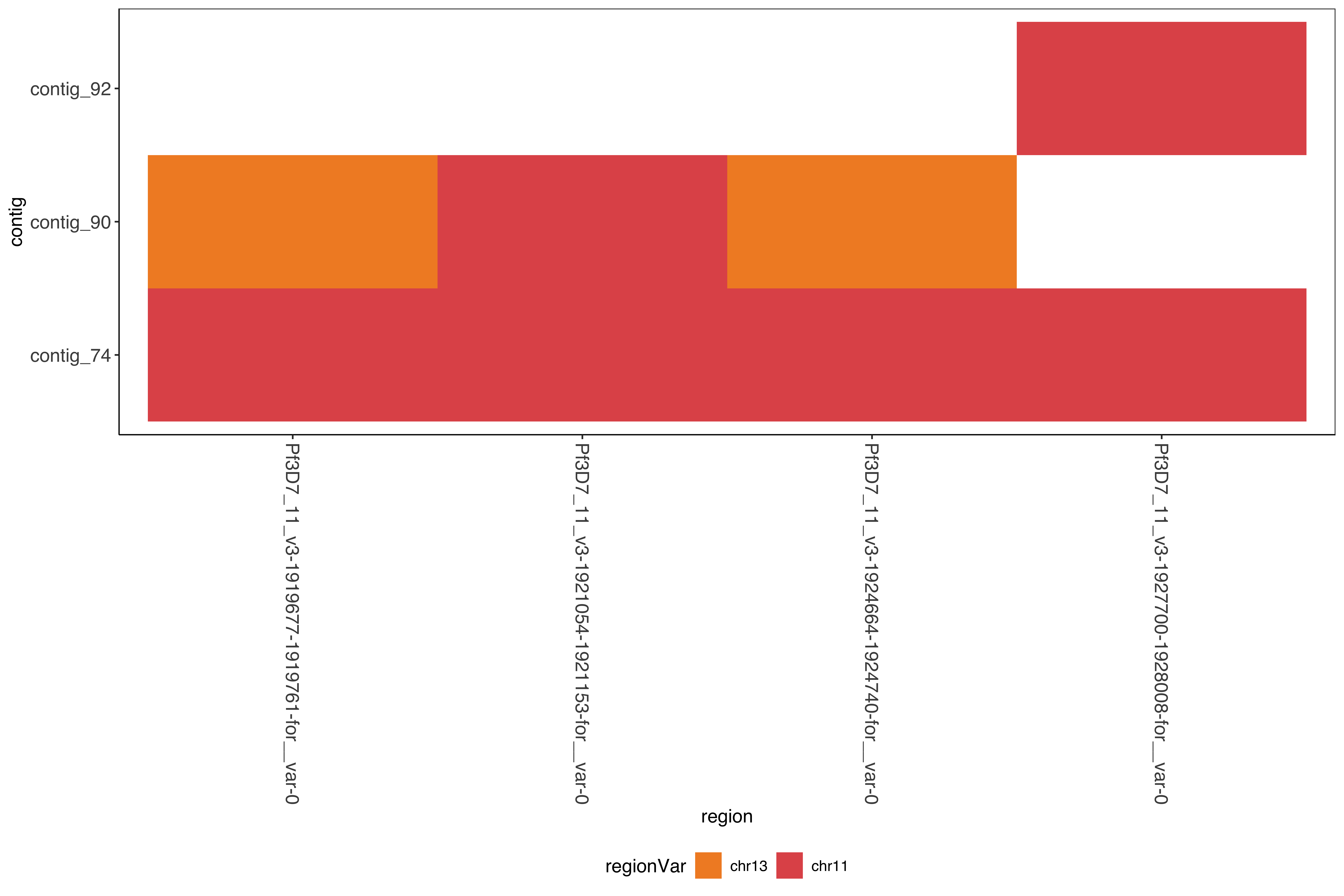
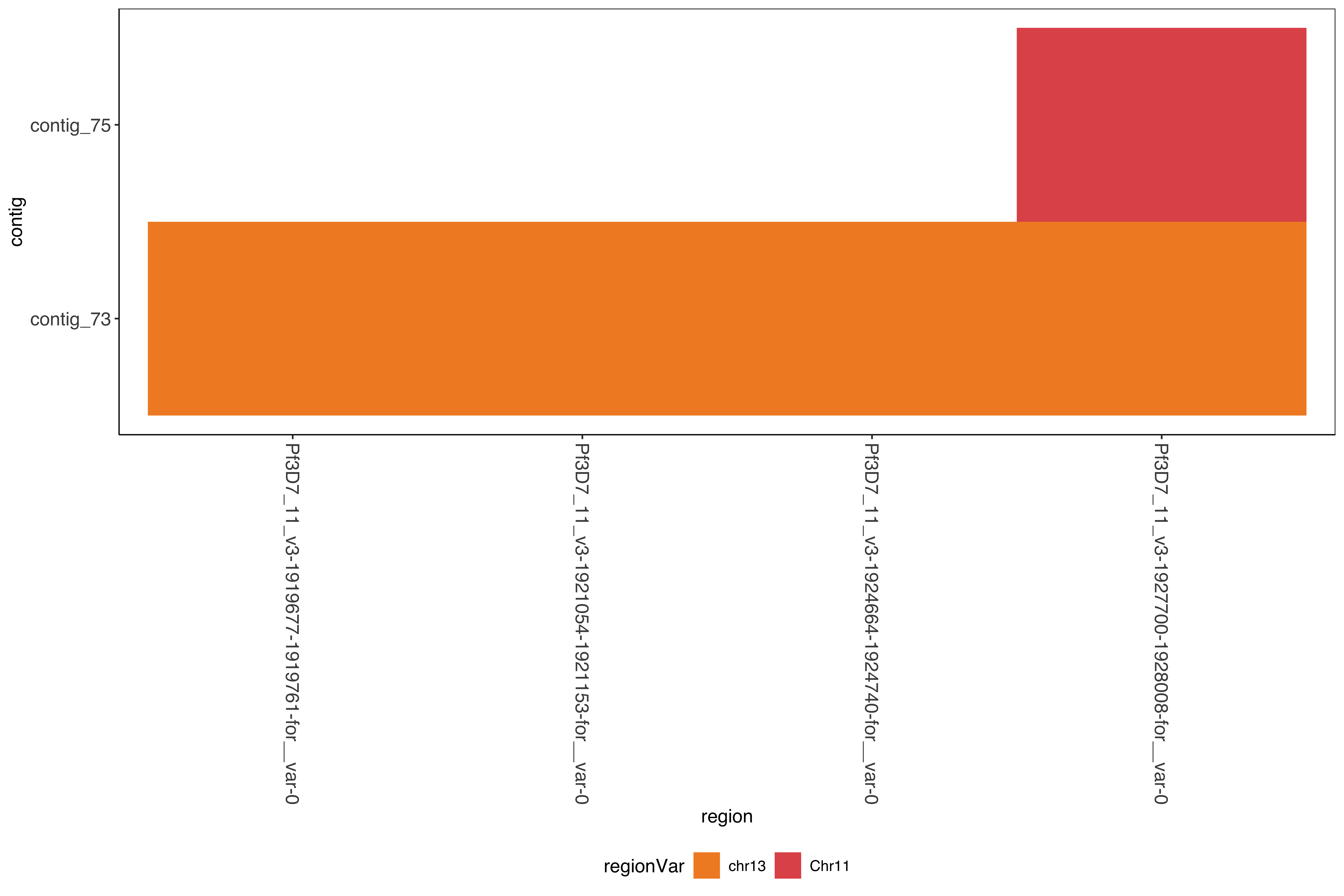
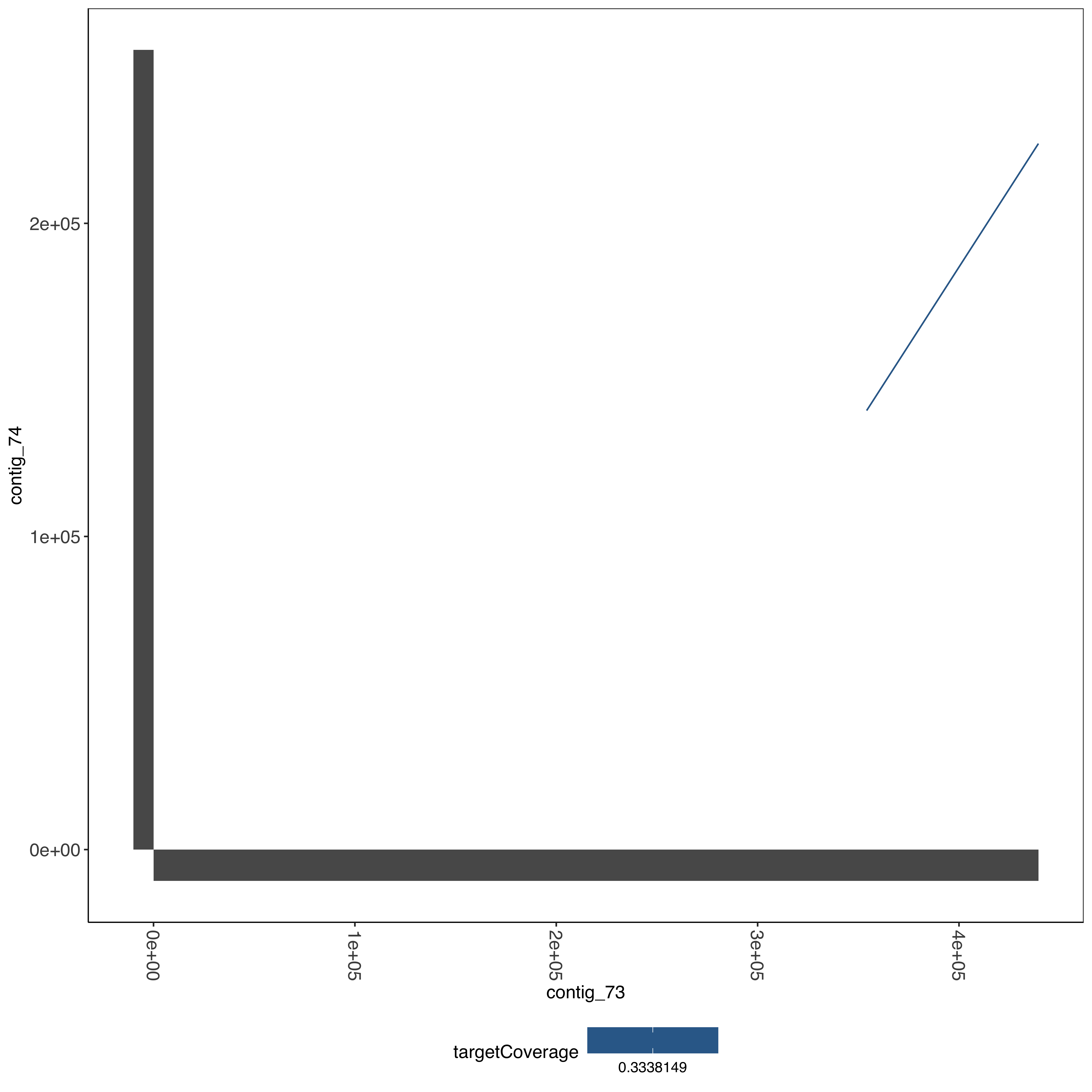
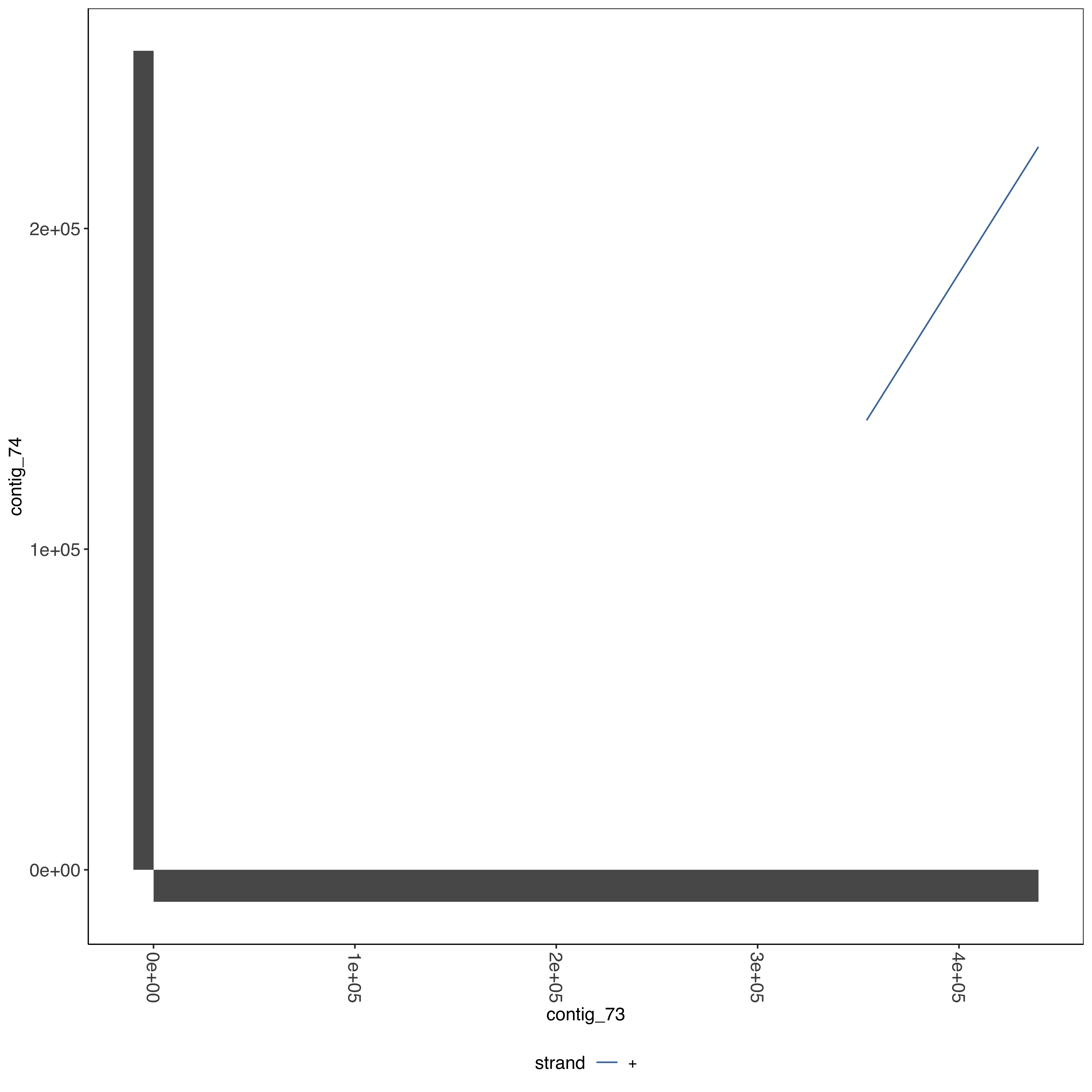
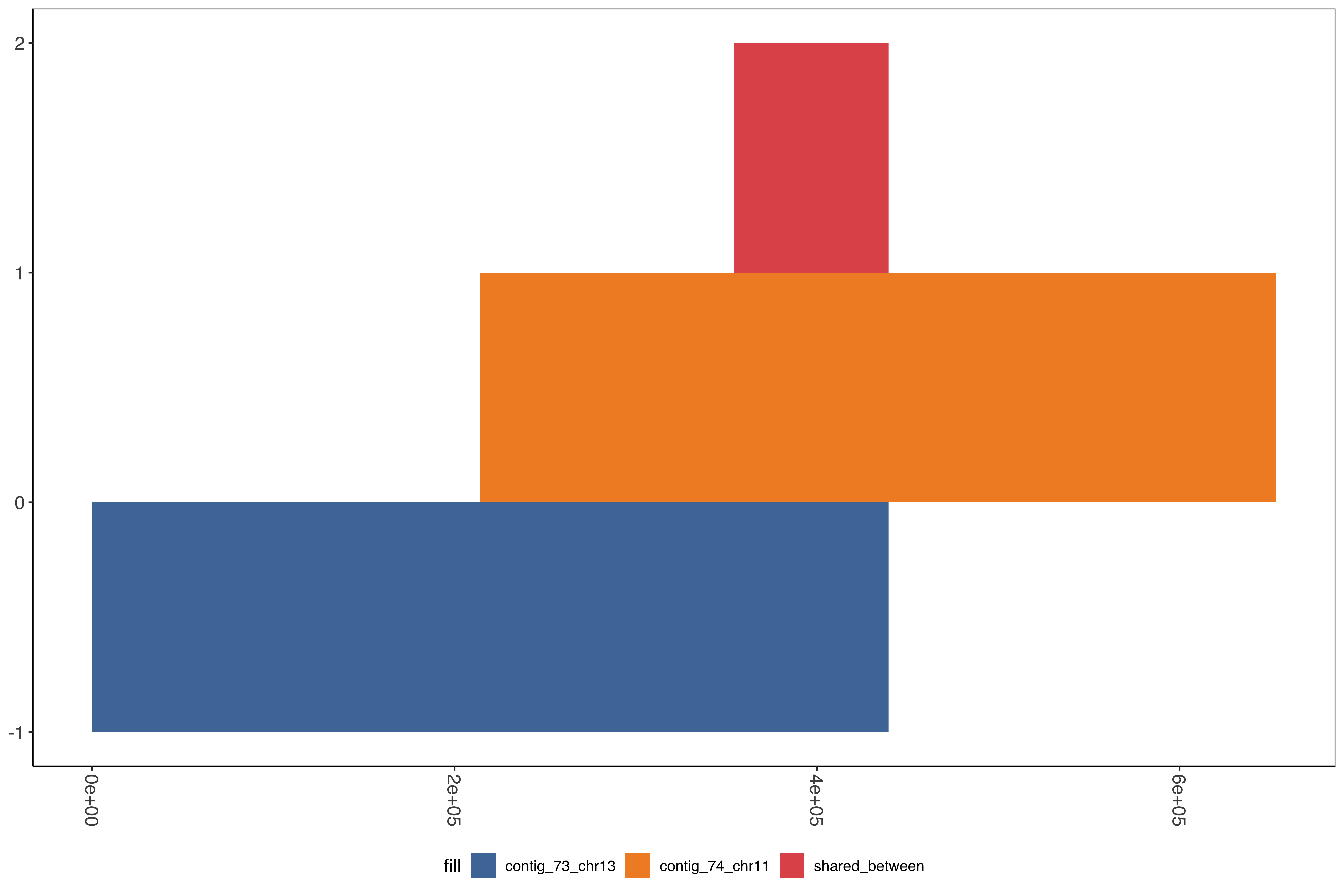
--- title: Running SD01 assemblies --- ```{r setup, echo=FALSE, message=FALSE} source("../common.R") ``` [ @Kolmogorov2019-lg ] assemblies on nanopore of SD01. * Running on all reads * Assembly on reads with chromosome 11 specific variation removed * Assembly on reads with chromosome 13 specific variation removed ```{bash, eval = F} cd /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore nohup flye --threads 44 --nano-raw rawFastq/PfSD01PermethION.fastq.gz --out-dir ownAssemblies/flyeAssemblyDefault & nohup flye --threads 44 --nano-raw extractingFromRawFastqs/nano_allButChr13_PfSD01PermethION.fastq.gz --out-dir ownAssemblies/flyeAssemblyDefaultForChr11 & nohup flye --threads 44 --nano-raw extractingFromRawFastqs/nano_allButChr11_PfSD01PermethION.fastq.gz --out-dir ownAssemblies/flyeAssemblyDefaultForChr13 & ``` ```{bash, eval = F} cd ownAssemblies mkdir nucmerResults minimap2Results # default flye nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta flyeAssemblyDefault/assembly.fasta --prefix nucmerResults/PfSD01_defaultflye_to_PfSD01_nucmer show-coords -T -l -c -H nucmerResults/PfSD01_defaultflye_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_defaultflye_to_PfSD01_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_defaultflye_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_defaultflye_to_PfSD01_nucmer.delta.tsv nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta flyeAssemblyDefault/assembly.fasta --prefix nucmerResults/PfSD01_defaultflye_to_Pf3D7_nucmer show-coords -T -l -c -H nucmerResults/PfSD01_defaultflye_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_defaultflye_to_Pf3D7_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_defaultflye_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_defaultflye_to_Pf3D7_nucmer.delta.tsv nucmer /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta flyeAssemblyDefault/assembly.fasta --prefix nucmerResults/PfSD01_defaultflye_to_Pf3D7Hybrid_nucmer show-coords -T -l -c -H nucmerResults/PfSD01_defaultflye_to_Pf3D7Hybrid_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_defaultflye_to_Pf3D7Hybrid_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_defaultflye_to_Pf3D7Hybrid_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_defaultflye_to_Pf3D7Hybrid_nucmer.delta.tsv minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta flyeAssemblyDefault/assembly.fasta > minimap2Results/PfSD01_defaultflye_to_PfSD01.tab.txt minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta flyeAssemblyDefault/assembly.fasta > minimap2Results/PfSD01_defaultflye_to_Pf3D7.tab.txt minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta flyeAssemblyDefault/assembly.fasta > minimap2Results/PfSD01_defaultflye_to_Pf3D7Hybrid.tab.txt minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta flyeAssemblyDefault/assembly.fasta | samtools sort -o minimap2Results/PfSD01_defaultflye_to_PfSD01.sorted.bam && samtools index minimap2Results/PfSD01_defaultflye_to_PfSD01.sorted.bam minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta flyeAssemblyDefault/assembly.fasta | samtools sort -o minimap2Results/PfSD01_defaultflye_to_Pf3D7.sorted.bam && samtools index minimap2Results/PfSD01_defaultflye_to_Pf3D7.sorted.bam minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta flyeAssemblyDefault/assembly.fasta | samtools sort -o minimap2Results/PfSD01_defaultflye_to_Pf3D7Hybrid.sorted.bam && samtools index minimap2Results/PfSD01_defaultflye_to_Pf3D7Hybrid.sorted.bam # DefaultForChr11 nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta flyeAssemblyDefaultForChr11/assembly.fasta --prefix nucmerResults/PfSD01_DefaultFlyeForChr11_to_PfSD01_nucmer show-coords -T -l -c -H nucmerResults/PfSD01_DefaultFlyeForChr11_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr11_to_PfSD01_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_DefaultFlyeForChr11_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr11_to_PfSD01_nucmer.delta.tsv nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta flyeAssemblyDefaultForChr11/assembly.fasta --prefix nucmerResults/PfSD01_DefaultFlyeForChr11_to_Pf3D7_nucmer show-coords -T -l -c -H nucmerResults/PfSD01_DefaultFlyeForChr11_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr11_to_Pf3D7_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_DefaultFlyeForChr11_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr11_to_Pf3D7_nucmer.delta.tsv nucmer /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta flyeAssemblyDefaultForChr11/assembly.fasta --prefix nucmerResults/PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid_nucmer show-coords -T -l -c -H nucmerResults/PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid_nucmer.delta.tsv minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta flyeAssemblyDefaultForChr11/assembly.fasta > minimap2Results/PfSD01_DefaultFlyeForChr11_to_PfSD01.tab.txt minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta flyeAssemblyDefaultForChr11/assembly.fasta > minimap2Results/PfSD01_DefaultFlyeForChr11_to_Pf3D7.tab.txt minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta flyeAssemblyDefaultForChr11/assembly.fasta > minimap2Results/PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid.tab.txt minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta flyeAssemblyDefaultForChr11/assembly.fasta | samtools sort -o minimap2Results/PfSD01_DefaultFlyeForChr11_to_PfSD01.sorted.bam && samtools index minimap2Results/PfSD01_DefaultFlyeForChr11_to_PfSD01.sorted.bam minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta flyeAssemblyDefaultForChr11/assembly.fasta | samtools sort -o minimap2Results/PfSD01_DefaultFlyeForChr11_to_Pf3D7.sorted.bam && samtools index minimap2Results/PfSD01_DefaultFlyeForChr11_to_Pf3D7.sorted.bam minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta flyeAssemblyDefaultForChr11/assembly.fasta | samtools sort -o minimap2Results/PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid.sorted.bam && samtools index minimap2Results/PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid.sorted.bam # DefaultForChr13 nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta flyeAssemblyDefaultForChr13/assembly.fasta --prefix nucmerResults/PfSD01_DefaultFlyeForChr13_to_PfSD01_nucmer show-coords -T -l -c -H nucmerResults/PfSD01_DefaultFlyeForChr13_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr13_to_PfSD01_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_DefaultFlyeForChr13_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr13_to_PfSD01_nucmer.delta.tsv nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta flyeAssemblyDefaultForChr13/assembly.fasta --prefix nucmerResults/PfSD01_DefaultFlyeForChr13_to_Pf3D7_nucmer show-coords -T -l -c -H nucmerResults/PfSD01_DefaultFlyeForChr13_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr13_to_Pf3D7_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_DefaultFlyeForChr13_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr13_to_Pf3D7_nucmer.delta.tsv nucmer /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta flyeAssemblyDefaultForChr13/assembly.fasta --prefix nucmerResults/PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid_nucmer show-coords -T -l -c -H nucmerResults/PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid_nucmer.delta.tsv minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta flyeAssemblyDefaultForChr13/assembly.fasta > minimap2Results/PfSD01_DefaultFlyeForChr13_to_PfSD01.tab.txt minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta flyeAssemblyDefaultForChr13/assembly.fasta > minimap2Results/PfSD01_DefaultFlyeForChr13_to_Pf3D7.tab.txt minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta flyeAssemblyDefaultForChr13/assembly.fasta > minimap2Results/PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid.tab.txt minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta flyeAssemblyDefaultForChr13/assembly.fasta | samtools sort -o minimap2Results/PfSD01_DefaultFlyeForChr13_to_PfSD01.sorted.bam && samtools index minimap2Results/PfSD01_DefaultFlyeForChr13_to_PfSD01.sorted.bam minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta flyeAssemblyDefaultForChr13/assembly.fasta | samtools sort -o minimap2Results/PfSD01_DefaultFlyeForChr13_to_Pf3D7.sorted.bam && samtools index minimap2Results/PfSD01_DefaultFlyeForChr13_to_Pf3D7.sorted.bam minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta flyeAssemblyDefaultForChr13/assembly.fasta | samtools sort -o minimap2Results/PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid.sorted.bam && samtools index minimap2Results/PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid.sorted.bam ``` ```{bash, eval = F} cd /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/PfSD01_withSD01MultiInShared_extractions_pureHybrid/PfSD01_combined_finalHrpSubwindows_regions_withSD01MultiInShared_inPureHybrid_regions elucidator extractFromGenomesAndCompare --genomeDir /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/ownAssemblies/flyeAssemblyDefault/ --numThreads 20 --target multiInShared --fastq all.fastq --program krush --sample PfSD01 --verbose --overWriteDir --dout compAgainstAssemblies_flyeDefaultAssembly elucidator extractFromGenomesAndCompare --genomeDir /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/ownAssemblies/flyeAssemblyDefaultForChr11/ --numThreads 20 --target multiInShared --fastq all.fastq --program krush --sample PfSD01 --verbose --overWriteDir --dout compAgainstAssemblies_flyeDefaultForChr11 elucidator extractFromGenomesAndCompare --genomeDir /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/ownAssemblies/flyeAssemblyDefaultForChr13/ --numThreads 20 --target multiInShared --fastq all.fastq --program krush --sample PfSD01 --verbose --overWriteDir --dout compAgainstAssemblies_flyeDefaultForChr13 ``` ```{r} = readr:: read_tsv ("../../sharedBetween11_and_13/investigatingChrom11Chrom13/combined_shared_11_13_region.bed" , col_names = F)= sharedRegionWithHybrid %>% rename (target = X1, sharedStart = X2, sharedEnd = X3) %>% select (target, sharedStart, sharedEnd)``` ## Minimap2 output columns info ```{r} = c ("query" , "queryFullLen" , "queryStart" , "queryEnd" , "strand" , "target" , "targetFullLen" , "targetStart" , "targetEnd" , "basesMatched" , "totalBases" , "mappingQuality" )``` ## Default Flye assembly in shared region ```{r} = readr:: read_tsv ("ownAssemblies/minimap2Results/PfSD01_defaultflye_to_Pf3D7Hybrid.tab.txt" , col_names = F)colnames (PfSD01_defaultflye_to_Pf3D7Hybrid)[1 : length (minimap2ColNames)] = minimap2ColNames= PfSD01_defaultflye_to_Pf3D7Hybrid %>% mutate (querySubLen = queryEnd - queryStart) %>% mutate (queryCoverage = querySubLen/ queryFullLen) %>% mutate (targetSubLen = targetEnd - targetStart) %>% mutate (targetCoverage = targetSubLen/ targetFullLen) ``` ```{r} = PfSD01_defaultflye_to_Pf3D7Hybrid %>% filter (target %in% sharedRegionWithHybrid_region$ target) %>% left_join (sharedRegionWithHybrid_region) %>% filter ((targetStart < sharedStart & targetEnd > sharedStart) | < sharedEnd & targetEnd > sharedEnd)) %>% mutate (rowid = row_number ())create_dt (PfSD01_defaultflye_to_Pf3D7Hybrid_sharedRegion)``` ```{r} #| fig-column: screen-inset-shaded #| column: screen-inset-shaded ggplotly (ggplot () + geom_rect (aes (xmin = targetStart, xmax = targetEnd, ymin = rowid, ymax = rowid + 1 , fill = query, targetStart = targetStart, targetEnd = targetEnd, queryStart = queryStart, queryEnd = queryEnd,strand = strand, queryFullLen = queryFullLen,queryCoverage = queryCoverage), data = PfSD01_defaultflye_to_Pf3D7Hybrid_sharedRegion) + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 , ymax = 0 , start = start, end = end),fill = "#AA0A3C" ,data = sharedRegionWithHybrid %>% mutate (target = X1, start = X2, end = X3)) + + facet_wrap (~ target, scales = "free" ) + scale_fill_tableau ())``` ```{r} = readr:: read_tsv ("organized/compAgainstAssemblies_flyeDefaultAssembly/assembly/refComparisonInfo.tab.txt" )= sharedMultiCompTo_flyeDefaultAssembly %>% mutate (region = gsub (" \\ ..*" , "" , ReadId)) %>% mutate (contig = gsub ("-.*" , "" , BestRef)) %>% mutate (regionVar = gsub (".*fastq." , "" , ReadId))= sharedMultiCompTo_flyeDefaultAssembly%>% group_by (region, contig) %>% mutate (maxScore = max (score)) %>% mutate (isMaxScore = maxScore == score)%>% filter (score == max (score)) %>% arrange (contig) %>% filter (hqScore > 0.925 ) %>% group_by ()create_dt (sharedMultiCompTo_flyeDefaultAssembly_best)ggplot (sharedMultiCompTo_flyeDefaultAssembly_best) + geom_tile (aes (x = region, y = contig, fill = regionVar)) + + scale_fill_manual (values = c ("0" = "#F28D2C" , "1" = "#E15758" ), labels = c ("chr13" , "chr11" ))``` ## Default Flye assembly trying for Chr11 ### in shared region ```{r} = readr:: read_tsv ("ownAssemblies/minimap2Results/PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid.tab.txt" , col_names = F)colnames (PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid)[1 : length (minimap2ColNames)] = minimap2ColNames= PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid %>% mutate (querySubLen = queryEnd - queryStart) %>% mutate (queryCoverage = querySubLen/ queryFullLen) %>% mutate (targetSubLen = targetEnd - targetStart) %>% mutate (targetCoverage = targetSubLen/ targetFullLen) ``` ```{r} = PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid %>% filter (target %in% sharedRegionWithHybrid_region$ target) %>% left_join (sharedRegionWithHybrid_region) %>% filter ((targetStart < sharedStart & targetEnd > sharedStart) | < sharedEnd & targetEnd > sharedEnd)) %>% mutate (rowid = row_number ())create_dt (PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid_sharedRegion)``` ```{r} #| fig-column: screen-inset-shaded #| column: screen-inset-shaded ggplotly (ggplot () + geom_rect (aes (xmin = targetStart, xmax = targetEnd, ymin = rowid, ymax = rowid + 1 , fill = query, targetStart = targetStart, targetEnd = targetEnd, queryStart = queryStart, queryEnd = queryEnd,strand = strand, queryFullLen = queryFullLen,queryCoverage = queryCoverage), data = PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid_sharedRegion) + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 , ymax = 0 , start = start, end = end),fill = "#AA0A3C" ,data = sharedRegionWithHybrid %>% mutate (target = X1, start = X2, end = X3)) + + facet_wrap (~ target, scales = "free" ) + scale_fill_tableau ())``` ```{r} = readr:: read_tsv ("organized/compAgainstAssemblies_flyeDefaultForChr11/assembly/refComparisonInfo.tab.txt" )= sharedMultiCompTo_flyeDefaultForChr11 %>% mutate (region = gsub (" \\ ..*" , "" , ReadId)) %>% mutate (contig = gsub ("-.*" , "" , BestRef)) %>% mutate (regionVar = gsub (".*fastq." , "" , ReadId))= sharedMultiCompTo_flyeDefaultForChr11%>% group_by (region, contig) %>% mutate (maxScore = max (score)) %>% mutate (isMaxScore = maxScore == score)%>% filter (score == max (score)) %>% arrange (contig) %>% filter (hqScore > 0.925 ) %>% group_by ()create_dt (sharedMultiCompTo_flyeDefaultForChr11_best)``` ```{r} ggplot (sharedMultiCompTo_flyeDefaultForChr11_best) + geom_tile (aes (x = region, y = contig, fill = regionVar)) + + scale_fill_manual (values = c ("0" = "#F28D2C" , "1" = "#E15758" ), labels = c ("chr13" , "chr11" ))``` ### In shared region and beyond ```{r} = PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid %>% filter (target %in% sharedRegionWithHybrid_region$ target) %>% left_join (sharedRegionWithHybrid_region) %>% filter ((targetStart < sharedStart & targetEnd > sharedStart) | >= sharedStart)) %>% filter (querySubLen > 1000 , queryCoverage > 0.50 ) %>% mutate (rowid = row_number ())create_dt (PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid_sharedRegionAndAfter)``` ```{r} #| fig-column: screen-inset-shaded #| column: screen-inset-shaded ggplotly (ggplot () + geom_rect (aes (xmin = targetStart, xmax = targetEnd, ymin = rowid, ymax = rowid + 1 , fill = query, targetStart = targetStart, targetEnd = targetEnd, queryStart = queryStart, queryEnd = queryEnd,strand = strand, queryFullLen = queryFullLen,queryCoverage = queryCoverage), data = PfSD01_DefaultFlyeForChr11_to_Pf3D7Hybrid_sharedRegionAndAfter) + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 , ymax = 0 , start = start, end = end),fill = "#AA0A3C" ,data = sharedRegionWithHybrid %>% mutate (target = X1, start = X2, end = X3)) + + facet_wrap (~ target, scales = "free" ) + scale_fill_tableau (palette = "Tableau 20" ))``` ## Default Flye assembly trying for Chr13 ### in shared region ```{r} = readr:: read_tsv ("ownAssemblies/minimap2Results/PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid.tab.txt" , col_names = F)colnames (PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid)[1 : length (minimap2ColNames)] = minimap2ColNames= PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid %>% mutate (querySubLen = queryEnd - queryStart) %>% mutate (queryCoverage = querySubLen/ queryFullLen) %>% mutate (targetSubLen = targetEnd - targetStart) %>% mutate (targetCoverage = targetSubLen/ targetFullLen) ``` ```{r} = PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid %>% filter (target %in% sharedRegionWithHybrid_region$ target) %>% left_join (sharedRegionWithHybrid_region) %>% filter ((targetStart < (sharedStart-1000 ) & targetEnd > (sharedStart-1000 )) | < sharedEnd & targetEnd > sharedEnd)) %>% mutate (rowid = row_number ())create_dt (PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid_sharedRegion)``` ```{r} #| fig-column: screen-inset-shaded #| column: screen-inset-shaded ggplotly (ggplot () + geom_rect (aes (xmin = targetStart, xmax = targetEnd, ymin = rowid, ymax = rowid + 1 , fill = query, targetStart = targetStart, targetEnd = targetEnd, queryStart = queryStart, queryEnd = queryEnd,strand = strand, queryFullLen = queryFullLen,queryCoverage = queryCoverage), data = PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid_sharedRegion) + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 , ymax = 0 , start = start, end = end),fill = "#AA0A3C" ,data = sharedRegionWithHybrid %>% mutate (target = X1, start = X2, end = X3)) + + facet_wrap (~ target, scales = "free" ) + scale_fill_tableau ())``` ```{r} = readr:: read_tsv ("organized/compAgainstAssemblies_flyeDefaultForChr13/assembly/refComparisonInfo.tab.txt" )= sharedMultiCompTo_flyeDefaultForChr13 %>% mutate (region = gsub (" \\ ..*" , "" , ReadId)) %>% mutate (contig = gsub ("-.*" , "" , BestRef)) %>% mutate (regionVar = gsub (".*fastq." , "" , ReadId))= sharedMultiCompTo_flyeDefaultForChr13%>% group_by (region, contig) %>% mutate (maxScore = max (score)) %>% mutate (isMaxScore = maxScore == score)%>% filter (score == max (score)) %>% arrange (contig) %>% filter (hqScore > 0.925 ) %>% group_by ()create_dt (sharedMultiCompTo_flyeDefaultForChr13_best)``` ```{r} ggplot (sharedMultiCompTo_flyeDefaultForChr13_best) + geom_tile (aes (x = region, y = contig, fill = regionVar)) + + scale_fill_manual (values = c ("0" = "#F28D2C" , "1" = "#E15758" ), labels = c ("chr13" , "Chr11" ))``` ### In shared region and beyond ```{r} = PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid %>% filter (target %in% sharedRegionWithHybrid_region$ target) %>% left_join (sharedRegionWithHybrid_region) %>% filter ((targetStart < sharedStart & targetEnd > sharedStart) | >= sharedStart)) %>% filter (querySubLen > 1000 , queryCoverage > 0.50 ) %>% mutate (rowid = row_number ())create_dt (PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid_sharedRegionAndAfter)``` ```{r} #| fig-column: screen-inset-shaded #| column: screen-inset-shaded ggplotly (ggplot () + geom_rect (aes (xmin = targetStart, xmax = targetEnd, ymin = rowid, ymax = rowid + 1 , fill = query, targetStart = targetStart, targetEnd = targetEnd, queryStart = queryStart, queryEnd = queryEnd,strand = strand, queryFullLen = queryFullLen,queryCoverage = queryCoverage), data = PfSD01_DefaultFlyeForChr13_to_Pf3D7Hybrid_sharedRegionAndAfter) + geom_rect (aes (xmin = X2, xmax = X3, ymin = - 10 , ymax = 0 , start = start, end = end),fill = "#AA0A3C" ,data = sharedRegionWithHybrid %>% mutate (target = X1, start = X2, end = X3)) + + facet_wrap (~ target, scales = "free" ) + scale_fill_tableau (palette = "Tableau 20" ))``` ## Combining ### Chromosome 11 ### Chromosome 13 ```{bash, eval = F} cd flyeAssemblyDefaultForChr11 elucidator reOrientReads --reOrientToBestWinner --fasta assembly.fasta --ref /tank/data/genomes/plasmodium//genomes/pf/genomes/Pf3D7.fasta --kLength 11 --overWrite --numThreads 40 sed -i 's/_Comp//g' reOriented_assembly.fasta ``` ```{bash, eval = F} cd flyeAssemblyDefaultForChr13 elucidator reOrientReads --reOrientToBestWinner --fasta assembly.fasta --ref /tank/data/genomes/plasmodium//genomes/pf/genomes/Pf3D7.fasta --kLength 11 --overWrite --numThreads 40 sed -i 's/_Comp//g' reOriented_assembly.fasta ``` ```{bash, eval = F} elucidator extractByName --fasta ../flyeAssemblyDefaultForChr11/reOriented_assembly.fasta --overWrite --out flyeAssemblyDefaultForChr11_contig_74 --names contig_74 elucidator extractByName --fasta ../flyeAssemblyDefaultForChr11/reOriented_assembly.fasta --overWrite --out flyeAssemblyDefaultForChr11_no_contig_90 --names contig_90 --excluding elucidator extractByName --fasta ../flyeAssemblyDefaultForChr13/reOriented_assembly.fasta --overWrite --out flyeAssemblyDefaultForChr13_contig_73 --names contig_73 elucidator extractByName --fasta ../flyeAssemblyDefaultForChr13/reOriented_assembly.fasta --overWrite --out flyeAssemblyDefaultForChr13_no_contig_209 --names contig_209 --excluding ``` ### comparing the two spanning contigs ```{bash, eval = F} # comp the two mkdir nucmerResults minimap2Results nucmer flyeAssemblyDefaultForChr11_contig_74.fasta flyeAssemblyDefaultForChr13_contig_73.fasta --prefix nucmerResults/ForChr13_contig_73_to_ForChr11_contig_74_nucmer show-coords -T -l -c -H nucmerResults/ForChr13_contig_73_to_ForChr11_contig_74_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/ForChr13_contig_73_to_ForChr11_contig_74_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/ForChr13_contig_73_to_ForChr11_contig_74_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/ForChr13_contig_73_to_ForChr11_contig_74_nucmer.delta.tsv minimap2 -t 44 -x asm5 flyeAssemblyDefaultForChr11_contig_74.fasta flyeAssemblyDefaultForChr13_contig_73.fasta > minimap2Results/ForChr13_contig_73_to_ForChr11_contig_74.tab.txt ``` ```{r} = readr:: read_tsv ("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/minimap2Results/ForChr13_contig_73_to_ForChr11_contig_74.tab.txt" , col_names = F)colnames (ForChr13_contig_73_to_ForChr11_contig_74)[1 : length (minimap2ColNames)] = minimap2ColNames= ForChr13_contig_73_to_ForChr11_contig_74 %>% mutate (querySubLen = queryEnd - queryStart) %>% mutate (queryCoverage = querySubLen/ queryFullLen) %>% mutate (targetSubLen = targetEnd - targetStart) %>% mutate (targetCoverage = targetSubLen/ targetFullLen) %>% filter (basesMatched > 200 )= ForChr13_contig_73_to_ForChr11_contig_74 %>% select (target, targetFullLen) %>% unique ()= ForChr13_contig_73_to_ForChr11_contig_74 %>% select (query, queryFullLen) %>% unique ()= ggplot () + geom_segment (aes (x = queryStart, xend = queryEnd, y = targetStart, yend = targetEnd, querySubLen = querySubLen,targetSubLen = targetSubLen,query = query, target = target, color = targetCoverage,targetCoverage = targetCoverage, queryCoverage = queryCoveragedata = ForChr13_contig_73_to_ForChr11_contig_74 %>% filter (targetSubLen > 500 )) + geom_rect (aes (ymin = - 10000 , ymax = 0 , xmin = 0 , xmax = queryFullLen, query = query, queryFullLen = queryFullLen),data = ForChr13_contig_73_to_ForChr11_contig_74_queryInfo + geom_rect (aes (xmin = - 10000 , xmax = 0 , ymin = 0 , ymax = targetFullLen, target = target, targetFullLen = targetFullLen),data = ForChr13_contig_73_to_ForChr11_contig_74_targetInfo+ labs (x = ForChr13_contig_73_to_ForChr11_contig_74_queryInfo$ query, y = ForChr13_contig_73_to_ForChr11_contig_74_targetInfo$ target) + ``` #### Minimap2 plot ```{r} #| column: screen-inset-shaded #| fig-column: screen-inset-shaded #| fig-width: 10 #| fig-height: 10 print (ForChr13_contig_73_to_ForChr11_contig_74_plot)``` ```{r} #| column: screen-inset-shaded #| fig-column: screen-inset-shaded #| fig-width: 10 #| fig-height: 10 ggplotly (ForChr13_contig_73_to_ForChr11_contig_74_plot)``` ```{r} = readr:: read_tsv ("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/nucmerResults/ForChr13_contig_73_to_ForChr11_contig_74_nucmer.delta.tsv" )= ForChr13_contig_73_to_ForChr11_contig_74_nuc %>% mutate (targetStart = col.1 , targetEnd = col.2 , target = col.0 , query = col.3 , length = col.4 , strand = col.5 )= ggplot () + geom_segment (aes (x = queryStart, xend = queryEnd, y = targetStart, yend = targetEnd, length = length,query = query, target = target, color = strand,perID = perID,strand = stranddata = ForChr13_contig_73_to_ForChr11_contig_74_nuc %>% filter (length > 500 )) + geom_rect (aes (ymin = - 10000 , ymax = 0 , xmin = 0 , xmax = queryFullLen, query = query, queryFullLen = queryFullLen),data = ForChr13_contig_73_to_ForChr11_contig_74_queryInfo + geom_rect (aes (xmin = - 10000 , xmax = 0 , ymin = 0 , ymax = targetFullLen, target = target, targetFullLen = targetFullLen),data = ForChr13_contig_73_to_ForChr11_contig_74_targetInfo+ labs (x = ForChr13_contig_73_to_ForChr11_contig_74_queryInfo$ query, y = ForChr13_contig_73_to_ForChr11_contig_74_targetInfo$ target) + + scale_color_tableau ()``` #### nucmer plot ```{r} #| column: screen-inset-shaded #| fig-column: screen-inset-shaded #| fig-width: 10 #| fig-height: 10 print (ForChr13_contig_73_to_ForChr11_contig_74_nucplot)``` ```{r} #| column: screen-inset-shaded #| fig-column: screen-inset-shaded #| fig-width: 10 #| fig-height: 10 ggplotly (ForChr13_contig_73_to_ForChr11_contig_74_nucplot)``` #### Overlap plot ```{r} = ForChr13_contig_73_to_ForChr11_contig_74 %>% group_by (query) %>% slice_max (order = totalBases, n = 1 ) %>% mutate (relativeStart = queryStart - targetStart) %>% mutate (relativeEnd = relativeStart + queryFullLen)ggplot () + geom_rect (aes (ymin = 0 , ymax = - 1 , xmin = 0 , xmax = queryFullLen, query = query, queryFullLen = queryFullLen, fill = "contig_73_chr13" ),data = ForChr13_contig_73_to_ForChr11_contig_74_queryInfo + geom_rect (aes (ymin = 0 , ymax = 1 , xmin = relativeStart, xmax = relativeEnd, target = target, targetFullLen = targetFullLen, fill = "contig_74_chr11" ),data = ForChr13_contig_73_to_ForChr11_contig_74_queryInfo_relative + geom_rect (aes (xmin = queryStart, xmax = queryEnd, ymin = rowid , ymax = rowid + 1 , length = length,query = query, target = target, fill = "shared_between" ,strand = stranddata = ForChr13_contig_73_to_ForChr11_contig_74_nuc %>% filter (length > 300 ) %>% mutate (rowid = row_number ())) + + scale_fill_tableau ()``` ## Combine to make final contigs file ```{bash, eval = F} sed 's/contig_73/contig_73_chr13/g' flyeAssemblyDefaultForChr13_contig_73.fasta > renamed_flyeAssemblyDefaultForChr13_contig_73.fasta cat flyeAssemblyDefaultForChr11_no_contig_90.fasta renamed_flyeAssemblyDefaultForChr13_contig_73.fasta > combined.fasta ``` ### Polishing [ @Walker2014-rq ] ```{bash, eval = F} bwa mem -M -t 44 combined.fasta /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R1.fastq.gz /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R2.fastq.gz | samtools sort -@ 44 -o illuminaAgainstCombined.sorted.bam && samtools index illuminaAgainstCombined.sorted.bam exec java -Xmx160G -Xms20m -jar /home/linuxbrew/.linuxbrew/Cellar/pilon/1.23/pilon-1.23.jar --genome combined.fasta --jumps illuminaAgainstCombined.sorted.bam --threads 44 ``` ### Post polish checks [ @Walker2014-rq ] , recheck them again. ```{bash, eval = F} nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta pilon.fasta --prefix nucmerResults/pilon_to_PfSD01_nucmer show-coords -T -l -c -H nucmerResults/pilon_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/pilon_to_PfSD01_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/pilon_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/pilon_to_PfSD01_nucmer.delta.tsv nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta pilon.fasta --prefix nucmerResults/pilon_to_Pf3D7_nucmer show-coords -T -l -c -H nucmerResults/pilon_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/pilon_to_Pf3D7_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/pilon_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/pilon_to_Pf3D7_nucmer.delta.tsv nucmer /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta pilon.fasta --prefix nucmerResults/pilon_to_Pf3D7Hybrid_nucmer show-coords -T -l -c -H nucmerResults/pilon_to_Pf3D7Hybrid_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/pilon_to_Pf3D7Hybrid_nucmer.delta.bed elucidator splitColumnContainingMeta --file nucmerResults/pilon_to_Pf3D7Hybrid_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/pilon_to_Pf3D7Hybrid_nucmer.delta.tsv minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta pilon.fasta > minimap2Results/pilon_to_PfSD01.tab.txt minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta pilon.fasta > minimap2Results/pilon_to_Pf3D7.tab.txt minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta pilon.fasta > minimap2Results/pilon_to_Pf3D7Hybrid.tab.txt minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta pilon.fasta | samtools sort -o minimap2Results/pilon_to_PfSD01.sorted.bam && samtools index minimap2Results/pilon_to_PfSD01.sorted.bam minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta pilon.fasta | samtools sort -o minimap2Results/pilon_to_Pf3D7.sorted.bam && samtools index minimap2Results/pilon_to_Pf3D7.sorted.bam minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta pilon.fasta | samtools sort -o minimap2Results/pilon_to_Pf3D7Hybrid.sorted.bam && samtools index minimap2Results/pilon_to_Pf3D7Hybrid.sorted.bam bwa mem -M -t 44 pilon.fasta /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R1.fastq.gz /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R2.fastq.gz | samtools sort -@ 44 -o illuminaAgainstPilon.sorted.bam && samtools index illuminaAgainstPilon.sorted.bam ``` ```{bash, eval = F} cd /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/PfSD01_withSD01MultiInShared_extractions_pureHybrid/PfSD01_combined_finalHrpSubwindows_regions_withSD01MultiInShared_inPureHybrid_regions elucidator extractFromGenomesAndCompare --genomeDir //tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/ --numThreads 20 --target multiInShared --fastq all.fastq --program krush --sample PfSD01 --verbose --overWriteDir --dout compAgainstAssemblies_combinedPilon --genomes pilon ``` ### Mapping polished final contigs against PfSD01 ```{r} #| column: screen-inset-shaded = readr:: read_tsv ("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/minimap2Results/pilon_to_PfSD01.tab.txt" )colnames (pilon_to_PfSD01)[1 : length (minimap2ColNames)] = minimap2ColNames= pilon_to_PfSD01 %>% mutate (querySubLen = queryEnd - queryStart) %>% mutate (queryCoverage = querySubLen/ queryFullLen) %>% mutate (targetSubLen = targetEnd - targetStart) %>% mutate (targetCoverage = targetSubLen/ targetFullLen) %>% group_by (target) %>% arrange (targetStart) %>% mutate (rowid = row_number ()) = pilon_to_PfSD01 %>% select (target, targetFullLen) %>% unique () %>% arrange (targetFullLen)= list ()for (tar in pilon_to_PfSD01_targetInfo$ target) {= pilon_to_PfSD01 %>% filter (target == tar) %>% filter (queryCoverage > 0.05 ) %>% group_by (target) %>% arrange (targetStart) %>% mutate (rowid = row_number ()) %>% ungroup ()= pilon_to_PfSD01_targetInfo %>% filter (target == tar)if (nrow (pilon_to_PfSD01_tar) > 1 ) {= ggplot () + geom_rect (aes (xmin = targetStart,xmax = targetEnd,ymin = rowid,ymax = rowid + 1 ,targetStart = targetStart, targetEnd = targetEnd,query = query,queryCoverage = queryCoverage, fill = queryCoveragedata = pilon_to_PfSD01_tar+ geom_rect (aes (xmin = 0 ,xmax = targetFullLen,ymin = - 2 ,ymax = 0 fill = "#00A0FA" ,data = pilon_to_PfSD01_targetInfo_tar+ + labs (title = tar) + scale_fill_gradient (low = "#ffffb2" , high = "#e31a1c" )#targetPlots[[paste0(tar, "-h2")]] = htmltools::h2(tar) paste0 (tar, "-plot" )]] = ggplotly (pilon_to_PfSD01_tar_plot)"table" ]] = create_dt (pilon_to_PfSD01)# htmltools::tagList(targetPlots) ``` ```{r} #| results: asis #| echo: false cat (create_tabsetOfHtmlWidgets (targetPlots))``` ### Mapping polished final contigs against PfHybrid3D7 ```{r} #| column: screen-inset-shaded = readr:: read_tsv ("ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/minimap2Results/pilon_to_Pf3D7Hybrid.tab.txt" )colnames (pilon_to_Pf3D7Hybrid)[1 : length (minimap2ColNames)] = minimap2ColNames= pilon_to_Pf3D7Hybrid %>% mutate (querySubLen = queryEnd - queryStart) %>% mutate (queryCoverage = querySubLen/ queryFullLen) %>% mutate (targetSubLen = targetEnd - targetStart) %>% mutate (targetCoverage = targetSubLen/ targetFullLen) %>% group_by (target) %>% arrange (targetStart) %>% mutate (rowid = row_number ()) = pilon_to_Pf3D7Hybrid %>% select (target, targetFullLen) %>% unique () %>% arrange (targetFullLen)= list ()for (tar in pilon_to_Pf3D7Hybrid_targetInfo$ target) {= pilon_to_Pf3D7Hybrid %>% filter (target == tar) %>% filter (queryCoverage > 0.05 ) %>% group_by (target) %>% arrange (targetStart) %>% mutate (rowid = row_number ()) %>% ungroup ()= pilon_to_Pf3D7Hybrid_targetInfo %>% filter (target == tar)if (nrow (pilon_to_Pf3D7Hybrid_tar) > 1 ) {= ggplot () + geom_rect (aes (xmin = targetStart,xmax = targetEnd,ymin = rowid,ymax = rowid + 1 ,targetStart = targetStart, targetEnd = targetEnd,query = query,queryCoverage = queryCoverage, fill = queryCoveragedata = pilon_to_Pf3D7Hybrid_tar+ geom_rect (aes (xmin = 0 ,xmax = targetFullLen,ymin = - 2 ,ymax = 0 fill = "#00A0FA" ,data = pilon_to_Pf3D7Hybrid_targetInfo_tar+ + labs (title = tar) + scale_fill_gradient (low = "#ffffb2" , high = "#e31a1c" )#targetPlots[[paste0(tar, "-h2")]] = htmltools::h2(tar) paste0 (tar, "-plot" )]] = ggplotly (pilon_to_Pf3D7Hybrid_tar_plot)"table" ]] = create_dt (pilon_to_Pf3D7Hybrid)# htmltools::tagList(targetPlots) ``` ```{r} #| results: asis #| echo: false cat (create_tabsetOfHtmlWidgets (targetPlots))``` <!-- ## More polish --> <!-- Unclear if running polish a second time would be any better. --> <!-- ```{bash, eval = F} --> <!-- bwa mem -M -t 44 pilon.fasta /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R1.fastq.gz /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R2.fastq.gz | samtools sort -@ 44 -o illuminaAgainstPilon.sorted.bam && samtools index illuminaAgainstPilon.sorted.bam --> <!-- exec java -Xmx160G -Xms20m -jar /home/linuxbrew/.linuxbrew/Cellar/pilon/1.23/pilon-1.23.jar --genome pilon.fasta --jumps illuminaAgainstPilon.sorted.bam --threads 44 --output morePilon --> <!-- ``` --> <!-- # Canu --> <!-- Several attempts made to run canu assembler --> <!-- ```{bash, eval = F} --> <!-- canu -pacbio-raw extractingFromRawFastqs/pacbio_allButChr11_combined.fastq.gz -p PfSD01_pacbio_forchr13_canu -d ownAssemblies/PfSD01_pacbio_forchr13_canu genomeSize=23m stopOnLowCoverage=0 --> <!-- canu -nanopore-raw extractingFromRawFastqs/nano_allButChr11_PfSD01PermethION.fastq.gz -p PfSD01_nano_forchr13_canu -d ownAssemblies/PfSD01_nano_forchr13_canu genomeSize=23m stopOnLowCoverage=0 --> <!-- canu -pacbio-raw extractingFromRawFastqs/pacbio_allButChr11_combined.fastq.gz -nanopore-raw extractingFromRawFastqs/nano_allButChr11_PfSD01PermethION.fastq.gz -p PfSD01_pacbio_nano_forchr13_canu -d ownAssemblies/PfSD01_pacbio_nano_forchr13_canu genomeSize=23m stopOnLowCoverage=0 --> <!-- canu -pacbio-raw extractingFromRawFastqs/pacbio_allButChr13_combined.fastq.gz -p PfSD01_pacbio_forchr11_canu -d ownAssemblies/PfSD01_pacbio_forchr11_canu genomeSize=23m stopOnLowCoverage=0 --> <!-- canu -nanopore-raw extractingFromRawFastqs/nano_allButChr13_PfSD01PermethION.fastq.gz -p PfSD01_nano_forchr11_canu -d ownAssemblies/PfSD01_nano_forchr11_canu genomeSize=23m stopOnLowCoverage=0 --> <!-- canu -pacbio-raw extractingFromRawFastqs/pacbio_allButChr13_combined.fastq.gz -nanopore-raw extractingFromRawFastqs/nano_allButChr13_PfSD01PermethION.fastq.gz -p PfSD01_pacbio_nano_forchr11_canu -d ownAssemblies/PfSD01_pacbio_nano_forchr11_canu genomeSize=23m stopOnLowCoverage=0 --> <!-- canu -nanopore-raw rawFastq/PfSD01PermethION.fastq.gz -p PfSD01_nano_canu -d ownAssemblies/PfSD01_nano_canu genomeSize=23m stopOnLowCoverage=0 --> <!-- canu -pacbio-raw pacbioReads/combined.fastq.gz -p PfSD01_pacbio_canu -d ownAssemblies/PfSD01_nano_canu genomeSize=23m stopOnLowCoverage=0 --> <!-- canu -pacbio-raw pacbioReads/combined.fastq.gz -nanopore-raw rawFastq/PfSD01PermethION.fastq.gz -p PfSD01_pacbio_nano_canu -d ownAssemblies/PfSD01_pacbio_nano_canu genomeSize=23m stopOnLowCoverage=0 --> <!-- ``` --> <!-- ```{bash, eval = F} --> <!-- ## PfSD01_pacbio_nano_canu --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_nano_canu/PfSD01_pacbio_nano_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_nano_canu/PfSD01_pacbio_nano_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_nano_canu/PfSD01_pacbio_nano_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.tsv --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_nano_canu/PfSD01_pacbio_nano_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_nano_canu/PfSD01_pacbio_nano_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_nano_canu/PfSD01_pacbio_nano_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid.tab.txt --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_nano_canu/PfSD01_pacbio_nano_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_nano_canu/PfSD01_pacbio_nano_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_nano_canu/PfSD01_pacbio_nano_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid.sorted.bam --> <!-- ## PfSD01_pacbio_nano_forchr11_canu --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_nano_forchr11_canu/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_nano_forchr11_canu/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_nano_forchr11_canu/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.tsv --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_nano_forchr11_canu/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_nano_forchr11_canu/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_nano_forchr11_canu/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid.tab.txt --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_nano_forchr11_canu/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_nano_forchr11_canu/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_nano_forchr11_canu/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid.sorted.bam --> <!-- ## PfSD01_pacbio_nano_forchr13_canu --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_nano_forchr13_canu/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_nano_forchr13_canu/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_nano_forchr13_canu/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.tsv --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_nano_forchr13_canu/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_nano_forchr13_canu/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_nano_forchr13_canu/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid.tab.txt --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_nano_forchr13_canu/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_nano_forchr13_canu/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_nano_forchr13_canu/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid.sorted.bam --> <!-- ## PfSD01_pacbio_forchr11_canu --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_forchr11_canu/PfSD01_pacbio_forchr11_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_PfSD01_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_PfSD01_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_PfSD01_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_forchr11_canu/PfSD01_pacbio_forchr11_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_forchr11_canu/PfSD01_pacbio_forchr11_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.tsv --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_forchr11_canu/PfSD01_pacbio_forchr11_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_PfSD01.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_forchr11_canu/PfSD01_pacbio_forchr11_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_forchr11_canu/PfSD01_pacbio_forchr11_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid.tab.txt --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_forchr11_canu/PfSD01_pacbio_forchr11_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_PfSD01.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_PfSD01.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_forchr11_canu/PfSD01_pacbio_forchr11_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_forchr11_canu/PfSD01_pacbio_forchr11_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid.sorted.bam --> <!-- ## PfSD01_pacbio_forchr13_canu --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_forchr13_canu/PfSD01_pacbio_forchr13_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_PfSD01_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_PfSD01_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_PfSD01_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_forchr13_canu/PfSD01_pacbio_forchr13_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_forchr13_canu/PfSD01_pacbio_forchr13_canu.contigs.fasta --prefix nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_nucmer.delta.tsv --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_forchr13_canu/PfSD01_pacbio_forchr13_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_PfSD01.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_forchr13_canu/PfSD01_pacbio_forchr13_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_forchr13_canu/PfSD01_pacbio_forchr13_canu.contigs.fasta > minimap2Results/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid.tab.txt --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta PfSD01_pacbio_forchr13_canu/PfSD01_pacbio_forchr13_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_PfSD01.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_PfSD01.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta PfSD01_pacbio_forchr13_canu/PfSD01_pacbio_forchr13_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta PfSD01_pacbio_forchr13_canu/PfSD01_pacbio_forchr13_canu.contigs.fasta | samtools sort -o minimap2Results/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid.sorted.bam && samtools index minimap2Results/PfSD01_pacbio_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid.sorted.bam --> <!-- ``` --> <!-- ## PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01 --> <!-- ```{r} --> <!-- #| column: screen-inset-shaded --> <!-- PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01 = readr::read_tsv("ownAssemblies/minimap2Results/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid.tab.txt") --> <!-- colnames(PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01 = PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01 %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) --> <!-- PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_targetInfo = PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01 %>% --> <!-- select(target, targetFullLen) %>% --> <!-- unique() %>% --> <!-- arrange(targetFullLen) --> <!-- targetPlots = list() --> <!-- for(tar in PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_targetInfo$target) { --> <!-- PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_tar = PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01 %>% --> <!-- filter(target == tar) %>% --> <!-- filter(queryCoverage > 0.05) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) %>% --> <!-- ungroup() --> <!-- PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_targetInfo_tar = PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_targetInfo %>% --> <!-- filter(target == tar) --> <!-- if (nrow(PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_tar) > 1) { --> <!-- PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_tar_plot = ggplot() + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = targetStart, --> <!-- xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- query = query, --> <!-- queryCoverage = queryCoverage, --> <!-- fill = queryCoverage --> <!-- ), --> <!-- data = PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_tar --> <!-- ) + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = 0, --> <!-- xmax = targetFullLen, --> <!-- ymin = -2, --> <!-- ymax = 0 --> <!-- ), --> <!-- fill = "#00A0FA", --> <!-- data = PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_targetInfo_tar --> <!-- ) + --> <!-- sofonias_theme_xRotate_backgroundTransparent + --> <!-- labs(title = tar) + --> <!-- scale_fill_gradient(low = "#ffffb2", high = "#e31a1c") --> <!-- # targetPlots[[paste0(tar, "-h2")]] = htmltools::h2(tar) --> <!-- targetPlots[[paste0(tar, "-plot")]] = ggplotly(PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01_tar_plot) --> <!-- } --> <!-- } --> <!-- targetPlots[["table"]] = create_dt(PfSD01_pacbio_nano_canu.contigs.fasta_to_PfSD01) --> <!-- # htmltools::tagList(targetPlots) --> <!-- ``` --> <!-- :::: {.column-screen-inset} --> <!-- ```{r} --> <!-- #| results: asis --> <!-- #| echo: false --> <!-- cat(create_tabsetOfHtmlWidgets(targetPlots)) --> <!-- ``` --> <!-- :::: --> <!-- ### Plotting in shared region in hybrid --> <!-- ```{r} --> <!-- PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid = readr::read_tsv("ownAssemblies/minimap2Results/PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid.tab.txt", col_names = F) --> <!-- colnames(PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid = PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) --> <!-- ``` --> <!-- ```{r} --> <!-- PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid_sharedRegion = PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid %>% --> <!-- filter(target %in% sharedRegionWithHybrid_region$target) %>% --> <!-- left_join(sharedRegionWithHybrid_region) %>% --> <!-- filter((targetStart < (sharedStart-1000) & targetEnd > (sharedStart-1000)) | --> <!-- (targetStart < sharedEnd & targetEnd > sharedEnd)) %>% --> <!-- mutate(rowid = row_number()) --> <!-- create_dt(PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid_sharedRegion) --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen-inset-shaded --> <!-- #| column: screen-inset-shaded --> <!-- ggplotly(ggplot() + --> <!-- geom_rect(aes(xmin = targetStart, xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- fill = query, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- queryStart = queryStart, --> <!-- queryEnd = queryEnd, --> <!-- strand = strand, queryFullLen = queryFullLen, --> <!-- queryCoverage = queryCoverage), --> <!-- data = PfSD01_pacbio_nano_canu.contigs.fasta_to_Pf3D7Hybrid_sharedRegion) + --> <!-- geom_rect(aes(xmin = X2, xmax = X3, --> <!-- ymin = -10, --> <!-- ymax = 0, --> <!-- start = start, --> <!-- end = end), --> <!-- fill = "#AA0A3C", --> <!-- data = sharedRegionWithHybrid %>% --> <!-- mutate(target = X1, --> <!-- start = X2, --> <!-- end = X3)) + --> <!-- sofonias_theme + --> <!-- facet_wrap(~target, scales = "free") + --> <!-- scale_fill_tableau()) --> <!-- ``` --> <!-- ## PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01 --> <!-- ```{r} --> <!-- #| column: screen-inset-shaded --> <!-- PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01 = readr::read_tsv("ownAssemblies/minimap2Results/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01.tab.txt") --> <!-- colnames(PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01 = PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01 %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) --> <!-- PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_targetInfo = PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01 %>% --> <!-- select(target, targetFullLen) %>% --> <!-- unique() %>% --> <!-- arrange(targetFullLen) --> <!-- targetPlots = list() --> <!-- for(tar in PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_targetInfo$target) { --> <!-- PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_tar = PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01 %>% --> <!-- filter(target == tar) %>% --> <!-- filter(queryCoverage > 0.05) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) %>% --> <!-- ungroup() --> <!-- PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_targetInfo_tar = PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_targetInfo %>% --> <!-- filter(target == tar) --> <!-- if (nrow(PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_tar) > 1) { --> <!-- PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_tar_plot = ggplot() + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = targetStart, --> <!-- xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- query = query, --> <!-- queryCoverage = queryCoverage, --> <!-- fill = queryCoverage --> <!-- ), --> <!-- data = PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_tar --> <!-- ) + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = 0, --> <!-- xmax = targetFullLen, --> <!-- ymin = -2, --> <!-- ymax = 0 --> <!-- ), --> <!-- fill = "#00A0FA", --> <!-- data = PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_targetInfo_tar --> <!-- ) + --> <!-- sofonias_theme_xRotate_backgroundTransparent + --> <!-- labs(title = tar) + --> <!-- scale_fill_gradient(low = "#ffffb2", high = "#e31a1c") --> <!-- # targetPlots[[paste0(tar, "-h2")]] = htmltools::h2(tar) --> <!-- targetPlots[[paste0(tar, "-plot")]] = ggplotly(PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01_tar_plot) --> <!-- } --> <!-- } --> <!-- targetPlots[["table"]] = create_dt(PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_PfSD01) --> <!-- # htmltools::tagList(targetPlots) --> <!-- ``` --> <!-- :::: {.column-screen-inset} --> <!-- ```{r} --> <!-- #| results: asis --> <!-- #| echo: false --> <!-- cat(create_tabsetOfHtmlWidgets(targetPlots)) --> <!-- ``` --> <!-- :::: --> <!-- ### Plotting in shared region in hybrid --> <!-- ```{r} --> <!-- PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid = readr::read_tsv("ownAssemblies/minimap2Results/PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid.tab.txt", col_names = F) --> <!-- colnames(PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid = PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) --> <!-- ``` --> <!-- ```{r} --> <!-- PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_sharedRegion = PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid %>% --> <!-- filter(target %in% sharedRegionWithHybrid_region$target) %>% --> <!-- left_join(sharedRegionWithHybrid_region) %>% --> <!-- filter((targetStart < (sharedStart-1000) & targetEnd > (sharedStart-1000)) | --> <!-- (targetStart < sharedEnd & targetEnd > sharedEnd)) %>% --> <!-- mutate(rowid = row_number()) --> <!-- create_dt(PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_sharedRegion) --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen-inset-shaded --> <!-- #| column: screen-inset-shaded --> <!-- ggplotly(ggplot() + --> <!-- geom_rect(aes(xmin = targetStart, xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- fill = query, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- queryStart = queryStart, --> <!-- queryEnd = queryEnd, --> <!-- strand = strand, queryFullLen = queryFullLen, --> <!-- queryCoverage = queryCoverage), --> <!-- data = PfSD01_pacbio_nano_forchr11_canu.contigs.fasta_to_Pf3D7Hybrid_sharedRegion) + --> <!-- geom_rect(aes(xmin = X2, xmax = X3, --> <!-- ymin = -10, --> <!-- ymax = 0, --> <!-- start = start, --> <!-- end = end), --> <!-- fill = "#AA0A3C", --> <!-- data = sharedRegionWithHybrid %>% --> <!-- mutate(target = X1, --> <!-- start = X2, --> <!-- end = X3)) + --> <!-- sofonias_theme + --> <!-- facet_wrap(~target, scales = "free") + --> <!-- scale_fill_tableau()) --> <!-- ``` --> <!-- ## PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01 --> <!-- ```{r} --> <!-- #| column: screen-inset-shaded --> <!-- PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01 = readr::read_tsv("ownAssemblies/minimap2Results/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01.tab.txt") --> <!-- colnames(PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01 = PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01 %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) --> <!-- PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_targetInfo = PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01 %>% --> <!-- select(target, targetFullLen) %>% --> <!-- unique() %>% --> <!-- arrange(targetFullLen) --> <!-- targetPlots = list() --> <!-- for(tar in PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_targetInfo$target) { --> <!-- PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_tar = PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01 %>% --> <!-- filter(target == tar) %>% --> <!-- filter(queryCoverage > 0.05) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) %>% --> <!-- ungroup() --> <!-- PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_targetInfo_tar = PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_targetInfo %>% --> <!-- filter(target == tar) --> <!-- if (nrow(PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_tar) > 1) { --> <!-- PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_tar_plot = ggplot() + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = targetStart, --> <!-- xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- query = query, --> <!-- queryCoverage = queryCoverage, --> <!-- fill = queryCoverage --> <!-- ), --> <!-- data = PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_tar --> <!-- ) + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = 0, --> <!-- xmax = targetFullLen, --> <!-- ymin = -2, --> <!-- ymax = 0 --> <!-- ), --> <!-- fill = "#00A0FA", --> <!-- data = PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_targetInfo_tar --> <!-- ) + --> <!-- sofonias_theme_xRotate_backgroundTransparent + --> <!-- labs(title = tar) + --> <!-- scale_fill_gradient(low = "#ffffb2", high = "#e31a1c") --> <!-- # targetPlots[[paste0(tar, "-h2")]] = htmltools::h2(tar) --> <!-- targetPlots[[paste0(tar, "-plot")]] = ggplotly(PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01_tar_plot) --> <!-- } --> <!-- } --> <!-- targetPlots[["table"]] = create_dt(PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_PfSD01) --> <!-- # htmltools::tagList(targetPlots) --> <!-- ``` --> <!-- :::: {.column-screen-inset} --> <!-- ```{r} --> <!-- #| results: asis --> <!-- #| echo: false --> <!-- cat(create_tabsetOfHtmlWidgets(targetPlots)) --> <!-- ``` --> <!-- :::: --> <!-- ### Plotting in shared region in hybrid --> <!-- ```{r} --> <!-- PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid = readr::read_tsv("ownAssemblies/minimap2Results/PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid.tab.txt", col_names = F) --> <!-- colnames(PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid = PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) --> <!-- ``` --> <!-- ```{r} --> <!-- PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_sharedRegion = PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid %>% --> <!-- filter(target %in% sharedRegionWithHybrid_region$target) %>% --> <!-- left_join(sharedRegionWithHybrid_region) %>% --> <!-- filter((targetStart < (sharedStart-1000) & targetEnd > (sharedStart-1000)) | --> <!-- (targetStart < sharedEnd & targetEnd > sharedEnd)) %>% --> <!-- mutate(rowid = row_number()) --> <!-- create_dt(PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_sharedRegion) --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen-inset-shaded --> <!-- #| column: screen-inset-shaded --> <!-- ggplotly(ggplot() + --> <!-- geom_rect(aes(xmin = targetStart, xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- fill = query, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- queryStart = queryStart, --> <!-- queryEnd = queryEnd, --> <!-- strand = strand, queryFullLen = queryFullLen, --> <!-- queryCoverage = queryCoverage), --> <!-- data = PfSD01_pacbio_nano_forchr13_canu.contigs.fasta_to_Pf3D7Hybrid_sharedRegion) + --> <!-- geom_rect(aes(xmin = X2, xmax = X3, --> <!-- ymin = -10, --> <!-- ymax = 0, --> <!-- start = start, --> <!-- end = end), --> <!-- fill = "#AA0A3C", --> <!-- data = sharedRegionWithHybrid %>% --> <!-- mutate(target = X1, --> <!-- start = X2, --> <!-- end = X3)) + --> <!-- sofonias_theme + --> <!-- facet_wrap(~target, scales = "free") + --> <!-- scale_fill_tableau()) --> <!-- ``` --> <!-- ### Polishing --> <!-- #### PfSD01_pacbio_nano_canu --> <!-- First need to remove tig00000005 and tig00018355 to eliminate possibilities of compromising the good contigs from the pure nanopore assemblies --> <!-- Polish the assembly with pilon with the illumina reads to correct the final contigs using pilon[@Walker2014-rq] --> <!-- ```{bash, eval = F} --> <!-- cd ownAssemblies/PfSD01_pacbio_nano_canu --> <!-- elucidator printNames --fasta PfSD01_pacbio_nano_canu.contigs.fasta | egrep "reads=1\b" > toRemove.txt --> <!-- elucidator printNames --fasta PfSD01_pacbio_nano_canu.contigs.fasta | egrep tig00000005 >> toRemove.txt --> <!-- elucidator printNames --fasta PfSD01_pacbio_nano_canu.contigs.fasta | egrep tig00018355 >> toRemove.txt --> <!-- elucidator extractByName --fasta PfSD01_pacbio_nano_canu.contigs.fasta --names toRemove.txt --overWrite --excluding --out filtered_PfSD01_pacbio_nano_canu.contigs.fasta --> <!-- bwa mem -M -t 44 filtered_PfSD01_pacbio_nano_canu.contigs.fasta /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R1.fastq.gz /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R2.fastq.gz | samtools sort -@ 44 -o illuminaAgainstCanuPacbioNanoFiltered.sorted.bam && samtools index illuminaAgainstCanuPacbioNanoFiltered.sorted.bam --> <!-- exec java -Xmx160G -Xms20m -jar /home/linuxbrew/.linuxbrew/Cellar/pilon/1.23/pilon-1.23.jar --genome filtered_PfSD01_pacbio_nano_canu.contigs.fasta --jumps illuminaAgainstCanuPacbioNanoFiltered.sorted.bam --threads 44 --> <!-- ``` --> <!-- #### Merging --> <!-- ```{bash, eval = F} --> <!-- # merge with the illumina assembly, will hold off on that given the likelihood of failure around telomeres --> <!-- #merge_wrapper.py /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/ownAssemblies/PfSD01_pacbio_nano_canu/pilon.fasta /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/assemblies/unicycler_PfSD01/assembly.fasta -l 500 --> <!-- merge_wrapper.py /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/ownAssemblies/combining_flyeAssemblyDefaultForChr11_ForChr13/pilon.fasta /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/ownAssemblies/PfSD01_pacbio_nano_canu/pilon.fasta -l 500 --> <!-- ``` --> <!-- ##### post merge checks --> <!-- ```{bash, eval = F} --> <!-- mkdir nucmerResults minimap2Results --> <!-- # default flye --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta merged_out.fasta --prefix nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta merged_out.fasta --prefix nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta merged_out.fasta --prefix nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_nucmer.delta.tsv --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta merged_out.fasta > minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta merged_out.fasta > minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta merged_out.fasta > minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid.tab.txt --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta merged_out.fasta | samtools sort -o minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01.sorted.bam && samtools index minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta merged_out.fasta | samtools sort -o minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7.sorted.bam && samtools index minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta merged_out.fasta | samtools sort -o minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid.sorted.bam && samtools index minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid.sorted.bam --> <!-- ``` --> <!-- ##### Merged against PfSD01 --> <!-- ```{r} --> <!-- #| column: screen-inset-shaded --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01 = readr::read_tsv("ownAssemblies/PfSD01_pacbio_nano_canu_combinedWithNanoFlye/minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01.tab.txt") --> <!-- colnames(PfSD01_mergedCanuFlyePacbioNano_to_PfSD01)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01 = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01 %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_targetInfo = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01 %>% --> <!-- select(target, targetFullLen) %>% --> <!-- unique() %>% --> <!-- arrange(targetFullLen) --> <!-- targetPlots = list() --> <!-- for(tar in PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_targetInfo$target) { --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_tar = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01 %>% --> <!-- filter(target == tar) %>% --> <!-- filter(queryCoverage > 0.05) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) %>% --> <!-- ungroup() --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_targetInfo_tar = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_targetInfo %>% --> <!-- filter(target == tar) --> <!-- if (nrow(PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_tar) > 1) { --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_tar_plot = ggplot() + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = targetStart, --> <!-- xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- query = query, --> <!-- queryCoverage = queryCoverage, --> <!-- fill = queryCoverage --> <!-- ), --> <!-- data = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_tar --> <!-- ) + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = 0, --> <!-- xmax = targetFullLen, --> <!-- ymin = -2, --> <!-- ymax = 0 --> <!-- ), --> <!-- fill = "#00A0FA", --> <!-- data = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_targetInfo_tar --> <!-- ) + --> <!-- sofonias_theme_xRotate_backgroundTransparent + --> <!-- labs(title = tar) + --> <!-- scale_fill_gradient(low = "#ffffb2", high = "#e31a1c") --> <!-- # targetPlots[[paste0(tar, "-h2")]] = htmltools::h2(tar) --> <!-- targetPlots[[paste0(tar, "-plot")]] = ggplotly(PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_tar_plot) --> <!-- } --> <!-- } --> <!-- targetPlots[["table"]] = create_dt(PfSD01_mergedCanuFlyePacbioNano_to_PfSD01) --> <!-- # htmltools::tagList(targetPlots) --> <!-- ``` --> <!-- :::: {.column-screen-inset} --> <!-- ```{r} --> <!-- #| results: asis --> <!-- #| echo: false --> <!-- cat(create_tabsetOfHtmlWidgets(targetPlots)) --> <!-- ``` --> <!-- :::: --> <!-- ##### Merged against Pf3D7Hybrid --> <!-- ```{r} --> <!-- #| column: screen-inset-shaded --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid = readr::read_tsv("ownAssemblies/PfSD01_pacbio_nano_canu_combinedWithNanoFlye/minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid.tab.txt") --> <!-- colnames(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_targetInfo = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid %>% --> <!-- select(target, targetFullLen) %>% --> <!-- unique() %>% --> <!-- arrange(targetFullLen) --> <!-- targetPlots = list() --> <!-- for(tar in PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_targetInfo$target) { --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_tar = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid %>% --> <!-- filter(target == tar) %>% --> <!-- filter(queryCoverage > 0.05) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) %>% --> <!-- ungroup() --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_targetInfo_tar = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_targetInfo %>% --> <!-- filter(target == tar) --> <!-- if (nrow(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_tar) > 1) { --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_tar_plot = ggplot() + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = targetStart, --> <!-- xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- query = query, --> <!-- queryCoverage = queryCoverage, --> <!-- fill = queryCoverage --> <!-- ), --> <!-- data = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_tar --> <!-- ) + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = 0, --> <!-- xmax = targetFullLen, --> <!-- ymin = -2, --> <!-- ymax = 0 --> <!-- ), --> <!-- fill = "#00A0FA", --> <!-- data = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_targetInfo_tar --> <!-- ) + --> <!-- sofonias_theme_xRotate_backgroundTransparent + --> <!-- labs(title = tar) + --> <!-- scale_fill_gradient(low = "#ffffb2", high = "#e31a1c") --> <!-- # targetPlots[[paste0(tar, "-h2")]] = htmltools::h2(tar) --> <!-- targetPlots[[paste0(tar, "-plot")]] = ggplotly(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_tar_plot) --> <!-- } --> <!-- } --> <!-- targetPlots[["table"]] = create_dt(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid) --> <!-- # htmltools::tagList(targetPlots) --> <!-- ``` --> <!-- :::: {.column-screen-inset} --> <!-- ```{r} --> <!-- #| results: asis --> <!-- #| echo: false --> <!-- cat(create_tabsetOfHtmlWidgets(targetPlots)) --> <!-- ``` --> <!-- :::: --> <!-- ##### Plotting in shared region in hybrid --> <!-- ```{r} --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid = readr::read_tsv("ownAssemblies/PfSD01_pacbio_nano_canu_combinedWithNanoFlye/minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid.tab.txt", col_names = F) --> <!-- colnames(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) --> <!-- ``` --> <!-- ```{r} --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_sharedRegion = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid %>% --> <!-- filter(target %in% sharedRegionWithHybrid_region$target) %>% --> <!-- left_join(sharedRegionWithHybrid_region) %>% --> <!-- filter((targetStart < (sharedStart-1000) & targetEnd > (sharedStart-1000)) | --> <!-- (targetStart < sharedEnd & targetEnd > sharedEnd)) %>% --> <!-- mutate(rowid = row_number()) --> <!-- create_dt(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_sharedRegion) --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen-inset-shaded --> <!-- #| column: screen-inset-shaded --> <!-- ggplotly(ggplot() + --> <!-- geom_rect(aes(xmin = targetStart, xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- fill = query, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- queryStart = queryStart, --> <!-- queryEnd = queryEnd, --> <!-- strand = strand, queryFullLen = queryFullLen, --> <!-- queryCoverage = queryCoverage), --> <!-- data = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_sharedRegion) + --> <!-- geom_rect(aes(xmin = X2, xmax = X3, --> <!-- ymin = -10, --> <!-- ymax = 0, --> <!-- start = start, --> <!-- end = end), --> <!-- fill = "#AA0A3C", --> <!-- data = sharedRegionWithHybrid %>% --> <!-- mutate(target = X1, --> <!-- start = X2, --> <!-- end = X3)) + --> <!-- sofonias_theme + --> <!-- facet_wrap(~target, scales = "free") + --> <!-- scale_fill_tableau()) --> <!-- ``` --> <!-- ##### Merging with illumina assembly --> <!-- ```{bash, eval = F} --> <!-- # merge with the illumina assembly, will hold off on that given the likelihood of failure around telomeres --> <!-- #merge_wrapper.py /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/ownAssemblies/PfSD01_pacbio_nano_canu/pilon.fasta /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/assemblies/unicycler_PfSD01/assembly.fasta -l 500 --> <!-- mkdir mergingWithIllumina --> <!-- cd mergingWithIllumina --> <!-- merge_wrapper.py /tank/projects/plasmodium/falciparum/hrp/hrp3_deletion/nanopore/ownAssemblies/PfSD01_pacbio_nano_canu/merged_out.fasta /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/assemblies/unicycler_PfSD01/assembly.fasta -l 500 --> <!-- ``` --> <!-- ##### post merge checks --> <!-- ```{bash, eval = F} --> <!-- mkdir nucmerResults minimap2Results --> <!-- # default flye --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta merged_out.fasta --prefix nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta merged_out.fasta --prefix nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7_nucmer.delta.tsv --> <!-- nucmer /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta merged_out.fasta --prefix nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_nucmer --> <!-- show-coords -T -l -c -H nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_nucmer.delta | elucidator parseNucmerResultsToBed --coordsOutput STDIN --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_nucmer.delta.bed --> <!-- elucidator splitColumnContainingMeta --file nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_nucmer.delta.bed --delim tab --column col.6 --removeEmptyColumn --addHeader --overWrite --out nucmerResults/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_nucmer.delta.tsv --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta merged_out.fasta > minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta merged_out.fasta > minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7.tab.txt --> <!-- minimap2 -t 44 -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta merged_out.fasta > minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid.tab.txt --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/PfSD01.fasta merged_out.fasta | samtools sort -o minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01.sorted.bam && samtools index minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_plusPfSD01/genomes/Pf3D7.fasta merged_out.fasta | samtools sort -o minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7.sorted.bam && samtools index minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7.sorted.bam --> <!-- minimap2 -t 44 -a -x asm5 /tank/data/genomes/plasmodium/genomes/pf_hybrid/genomes/Pf3D7_plus_11-13_13-11_hybrid.fasta merged_out.fasta | samtools sort -o minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid.sorted.bam && samtools index minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid.sorted.bam --> <!-- ``` --> <!-- ##### Merged against PfSD01 --> <!-- ```{r} --> <!-- #| column: screen-inset-shaded --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01 = readr::read_tsv("ownAssemblies/PfSD01_pacbio_nano_canu_combinedWithNanoFlye/mergingWithIllumina/minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_PfSD01.tab.txt") --> <!-- colnames(PfSD01_mergedCanuFlyePacbioNano_to_PfSD01)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01 = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01 %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_targetInfo = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01 %>% --> <!-- select(target, targetFullLen) %>% --> <!-- unique() %>% --> <!-- arrange(targetFullLen) --> <!-- targetPlots = list() --> <!-- for(tar in PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_targetInfo$target) { --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_tar = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01 %>% --> <!-- filter(target == tar) %>% --> <!-- filter(queryCoverage > 0.05) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) %>% --> <!-- ungroup() --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_targetInfo_tar = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_targetInfo %>% --> <!-- filter(target == tar) --> <!-- if (nrow(PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_tar) > 1) { --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_tar_plot = ggplot() + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = targetStart, --> <!-- xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- query = query, --> <!-- queryCoverage = queryCoverage, --> <!-- fill = queryCoverage --> <!-- ), --> <!-- data = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_tar --> <!-- ) + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = 0, --> <!-- xmax = targetFullLen, --> <!-- ymin = -2, --> <!-- ymax = 0 --> <!-- ), --> <!-- fill = "#00A0FA", --> <!-- data = PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_targetInfo_tar --> <!-- ) + --> <!-- sofonias_theme_xRotate_backgroundTransparent + --> <!-- labs(title = tar) + --> <!-- scale_fill_gradient(low = "#ffffb2", high = "#e31a1c") --> <!-- # targetPlots[[paste0(tar, "-h2")]] = htmltools::h2(tar) --> <!-- targetPlots[[paste0(tar, "-plot")]] = ggplotly(PfSD01_mergedCanuFlyePacbioNano_to_PfSD01_tar_plot) --> <!-- } --> <!-- } --> <!-- targetPlots[["table"]] = create_dt(PfSD01_mergedCanuFlyePacbioNano_to_PfSD01) --> <!-- # htmltools::tagList(targetPlots) --> <!-- ``` --> <!-- :::: {.column-screen-inset} --> <!-- ```{r} --> <!-- #| results: asis --> <!-- #| echo: false --> <!-- cat(create_tabsetOfHtmlWidgets(targetPlots)) --> <!-- ``` --> <!-- :::: --> <!-- ##### Merged against Pf3D7Hybrid --> <!-- ```{r} --> <!-- #| column: screen-inset-shaded --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid = readr::read_tsv("ownAssemblies/PfSD01_pacbio_nano_canu_combinedWithNanoFlye/mergingWithIllumina/minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid.tab.txt") --> <!-- colnames(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_targetInfo = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid %>% --> <!-- select(target, targetFullLen) %>% --> <!-- unique() %>% --> <!-- arrange(targetFullLen) --> <!-- targetPlots = list() --> <!-- for(tar in PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_targetInfo$target) { --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_tar = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid %>% --> <!-- filter(target == tar) %>% --> <!-- filter(queryCoverage > 0.05) %>% --> <!-- group_by(target) %>% --> <!-- arrange(targetStart) %>% --> <!-- mutate(rowid = row_number()) %>% --> <!-- ungroup() --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_targetInfo_tar = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_targetInfo %>% --> <!-- filter(target == tar) --> <!-- if (nrow(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_tar) > 1) { --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_tar_plot = ggplot() + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = targetStart, --> <!-- xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- query = query, --> <!-- queryCoverage = queryCoverage, --> <!-- fill = queryCoverage --> <!-- ), --> <!-- data = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_tar --> <!-- ) + --> <!-- geom_rect( --> <!-- aes( --> <!-- xmin = 0, --> <!-- xmax = targetFullLen, --> <!-- ymin = -2, --> <!-- ymax = 0 --> <!-- ), --> <!-- fill = "#00A0FA", --> <!-- data = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_targetInfo_tar --> <!-- ) + --> <!-- sofonias_theme_xRotate_backgroundTransparent + --> <!-- labs(title = tar) + --> <!-- scale_fill_gradient(low = "#ffffb2", high = "#e31a1c") --> <!-- # targetPlots[[paste0(tar, "-h2")]] = htmltools::h2(tar) --> <!-- targetPlots[[paste0(tar, "-plot")]] = ggplotly(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_tar_plot) --> <!-- } --> <!-- } --> <!-- targetPlots[["table"]] = create_dt(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid) --> <!-- # htmltools::tagList(targetPlots) --> <!-- ``` --> <!-- :::: {.column-screen-inset} --> <!-- ```{r} --> <!-- #| results: asis --> <!-- #| echo: false --> <!-- cat(create_tabsetOfHtmlWidgets(targetPlots)) --> <!-- ``` --> <!-- :::: --> <!-- ##### Plotting in shared region in hybrid --> <!-- ```{r} --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid = readr::read_tsv("ownAssemblies/PfSD01_pacbio_nano_canu_combinedWithNanoFlye/mergingWithIllumina/minimap2Results/PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid.tab.txt", col_names = F) --> <!-- colnames(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid)[1:length(minimap2ColNames)] = minimap2ColNames --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid %>% --> <!-- mutate(querySubLen = queryEnd - queryStart) %>% --> <!-- mutate(queryCoverage = querySubLen/queryFullLen) %>% --> <!-- mutate(targetSubLen = targetEnd - targetStart) %>% --> <!-- mutate(targetCoverage = targetSubLen/targetFullLen) --> <!-- ``` --> <!-- ```{r} --> <!-- PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_sharedRegion = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid %>% --> <!-- filter(target %in% sharedRegionWithHybrid_region$target) %>% --> <!-- left_join(sharedRegionWithHybrid_region) %>% --> <!-- filter((targetStart < (sharedStart-1000) & targetEnd > (sharedStart-1000)) | --> <!-- (targetStart < sharedEnd & targetEnd > sharedEnd)) %>% --> <!-- mutate(rowid = row_number()) --> <!-- create_dt(PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_sharedRegion) --> <!-- ``` --> <!-- ```{r} --> <!-- #| fig-column: screen-inset-shaded --> <!-- #| column: screen-inset-shaded --> <!-- ggplotly(ggplot() + --> <!-- geom_rect(aes(xmin = targetStart, xmax = targetEnd, --> <!-- ymin = rowid, --> <!-- ymax = rowid + 1, --> <!-- fill = query, --> <!-- targetStart = targetStart, --> <!-- targetEnd = targetEnd, --> <!-- queryStart = queryStart, --> <!-- queryEnd = queryEnd, --> <!-- strand = strand, queryFullLen = queryFullLen, --> <!-- queryCoverage = queryCoverage), --> <!-- data = PfSD01_mergedCanuFlyePacbioNano_to_Pf3D7Hybrid_sharedRegion) + --> <!-- geom_rect(aes(xmin = X2, xmax = X3, --> <!-- ymin = -10, --> <!-- ymax = 0, --> <!-- start = start, --> <!-- end = end), --> <!-- fill = "#AA0A3C", --> <!-- data = sharedRegionWithHybrid %>% --> <!-- mutate(target = X1, --> <!-- start = X2, --> <!-- end = X3)) + --> <!-- sofonias_theme + --> <!-- facet_wrap(~target, scales = "free") + --> <!-- scale_fill_tableau()) --> <!-- ``` --> <!-- ## Unicycler --> <!-- Unicyler has a hybrid assembly method as well which I forget, will attempt --> <!-- ```{bash, eval = F} --> <!-- # ran --> <!-- unicycler -1 /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R1.fastq.gz -2 /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R2.fastq.gz -l rawFastq/PfSD01PermethION.fastq.gz -o ownAssemblies/SD01_unicycler_illumina_nanopore --> <!-- # pending --> <!-- unicycler -1 /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R1.fastq.gz -2 /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R2.fastq.gz -l rawFastq/PfSD01PermethION.fastq.gz -o ownAssemblies/SD01_unicycler_illumina_nanopore_bold --mode bold --> <!-- # pending --> <!-- unicycler -1 /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R1.fastq.gz -2 /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R2.fastq.gz -l pacbioReads/combined.fastq.gz -o ownAssemblies/SD01_unicycler_pacbio_nanopore --> <!-- # pending --> <!-- unicycler -1 /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R1.fastq.gz -2 /tank/data/plasmodium/falciparum/pfpubdata/WGS/reExtractedFastq/PfSD01_R2.fastq.gz -l pacbioReads/combined.fastq.gz -o ownAssemblies/SD01_unicycler_illumina_pacbio_bold --mode bold --> <!-- ``` --> <!-- ```{bash, eval = F} --> <!-- ``` --> <!-- ## Todo --> <!-- - [ ] map out canu assemblies especially with the pacbio assembly to see how they match up with the flye assembly --> <!-- - [ ] attempt to combine all assemblies (e.g. unicycler, canu with pacbio, and flye) --> <!-- - [ ] which would require polishing each genome assembly and then running merge_wrapper.py -->